簡介#
1928 年,Alexander Fleming 在倫敦聖瑪麗醫院觀察到青黴菌(Penicillium)可抑制金黃色葡萄球菌(staphylococci)的生長,並將此物質命名為盤尼西林(penicillin)。1940 年,Howard Florey 與 Ernst Chain 團隊在牛津進一步確認其強效抗菌與低毒性特性,開啟了「抗生素時代(antibiotic era)」。
細菌依據革蘭氏染色(Gram’s stain)可分為革蘭氏陽性菌(Gram-positive bacteria)與革蘭氏陰性菌(Gram-negative bacteria),兩者細胞壁結構差異顯著,直接影響抗生素的穿透性與療效:
- 革蘭氏陽性菌的細胞壁較厚(15–50 nm),主要由肽聚糖(peptidoglycan,約 50%)組成,帶強極性。
- 革蘭氏陰性菌的細胞壁更複雜,由周質空間(periplasmic space)、薄肽聚糖層(約 2 nm)、外膜(outer membrane)及脂多糖(lipopolysaccharide)組成;外膜上的孔蛋白(porins)可讓親水性抗生素通過,但整體屏障較強,使部分抗生素對其效力降低。
脂多糖是革蘭氏陰性菌對許多抗生素(如苄基青黴素、萬古黴素、利福平等)穿透的主要障礙,也是銅綠假單胞菌(Pseudomonas aeruginosa)表現驚人抗藥性的重要原因之一。
干擾葉酸合成或作用的抗微生物藥#
磺胺類藥物(Sulfonamides)#
磺胺類藥物於 1930 年代由 Gerhard Domagk 首先發現,代表性前驅藥為普朗托西(prontosil),在體內代謝為活性產物磺胺(sulfanilamide)。目前仍在使用的主要包括:
- 磺胺甲惡唑(sulfamethoxazole):通常與甲氧苄啶(trimethoprim)合用,即「複方磺胺甲惡唑(co-trimoxazole)」
- 柳氮磺吡啶(sulfasalazine):用於發炎性腸道病
- 磺胺嘧啶(sulfadiazine):偶爾使用
作用機制
磺胺類藥物是對胺基苯甲酸(p-aminobenzoic acid,PABA)的結構類似物,競爭性抑制細菌的二氫葉酸合成酶(dihydropteroate synthetase),阻斷葉酸(folate)合成,進而妨礙 DNA 與 RNA 的製造。磺胺類藥物屬於抑菌性(bacteriostatic),在膿液或組織壞死產物存在時,因細菌可直接利用胸腺嘧啶與嘌呤而使藥效減弱。
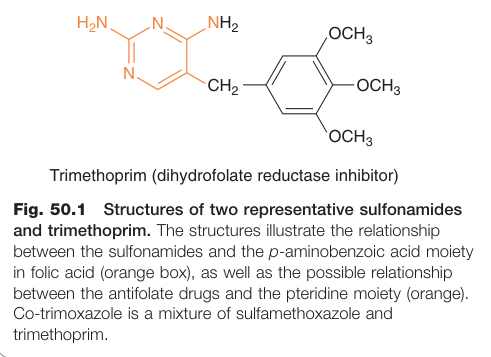
Figure 50.1:磺胺類藥物與甲氧苄啶的化學結構,顯示其與葉酸中 p-aminobenzoic acid 結構的關係
抗藥性(resistance)常見,由質粒(plasmid)介導,源於細菌產生對藥物不敏感的新酶。含 PABA 結構的局部麻醉藥(如普魯卡因)可拮抗其抗菌效果。
藥動學
- 口服吸收良好(柳氮磺吡啶除外)
- 可穿透胎盤與血腦屏障
- 主要在肝臟乙醯化代謝;乙醯化代謝物可在尿中沉澱導致結晶尿(crystalluria)
不良反應
- 嚴重:肝炎、骨髓抑制、急性腎衰竭、過敏反應(Stevens-Johnson syndrome、中毒性表皮壞死松解症、過敏性休克)
- 一般:噁心、嘔吐、頭痛、精神抑鬱、高鐵血紅蛋白血症所致發紺
臨床用途
- 與甲氧苄啶合用(co-trimoxazole)治療耶氏肺囊菌(P. jirovecii)肺炎
- 與乙胺嘧啶合用治療抗藥性瘧疾與弓漿蟲病(toxoplasmosis)
- 發炎性腸道病(柳氮磺吡啶)
- 燒傷感染(磺胺嘧啶銀局部使用)
- 諾卡氏菌病(nocardiasis)
甲氧苄啶(Trimethoprim)#
作用機制
甲氧苄啶結構上類似葉酸的蝶啶基(pteridine moiety),選擇性抑制細菌的二氫葉酸還原酶(dihydrofolate reductase),對人類同類酶的親和力遠低於細菌,具高度選擇性,屬抑菌性藥物。與磺胺類合用(co-trimoxazole)可在葉酸合成路徑的兩個不同節點發揮協同作用。
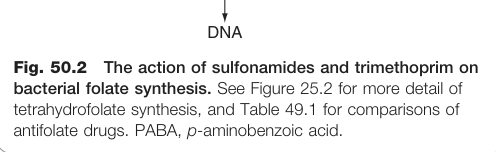
Figure 50.2:磺胺類藥物與甲氧苄啶作用於細菌葉酸合成路徑的兩個節點,顯示兩藥協同機制
藥動學
- 口服吸收良好,廣泛分布至組織與體液,包括肺、腎及腦脊液(CSF)
- 弱鹼性,尿液 pH 降低可加速腎臟排泄
- 約半數劑量在 24 小時內排出
不良反應
- 葉酸缺乏 → 巨紅血球性貧血(megaloblastic anaemia),可以亞葉酸(folinic acid)預防
- 噁心、嘔吐、皮疹及血液異常
臨床用途
- 單獨使用:泌尿道與呼吸道感染
- co-trimoxazole 高劑量:AIDS 患者的耶氏肺囊菌肺炎
β-內醯胺類抗生素(β-Lactam Antibiotics)#
所有 β-內醯胺類抗生素(β-lactams)的共同作用機制為干擾細菌細胞壁肽聚糖的合成:與細菌上的青黴素結合蛋白(penicillin-binding proteins,PBPs)結合後,抑制轉肽酶(transpeptidation enzyme)對肽聚糖骨架的交聯;最終透過活化自溶酶(autolytic enzymes)裂解細菌,屬於**殺菌性(bactericidal)**藥物。
β-內醯胺酶(β-lactamases)是細菌對此類藥物最主要的抗藥機制,可水解 β-內醯胺環使其失效。
青黴素類(Penicillins)#
種類與抗菌活性
| 類型 | 代表藥 | 特色 |
|---|---|---|
| 天然青黴素 | 苄基青黴素(penicillin G)、苯氧甲基青黴素(penicillin V) | 廣效,但需注射、易被 β-lactamase 破壞 |
| 耐 β-內醯胺酶型 | 氟氯西林(flucloxacillin)、替莫西林(temocillin) | 主用於葡萄球菌感染 |
| 廣效型 | 阿莫西林(amoxicillin)、氨苄西林(ampicillin) | 涵蓋革蘭氏陰性菌,但仍被 β-lactamase 破壞 |
| 抗假單胞菌型 | 哌拉西林(piperacillin)、替卡西林(ticarcillin) | 對銅綠假單胞菌有效 |
阿莫西林或替卡西林可與 β-內醯胺酶抑制劑**克拉維酸(clavulanic acid)**合用(如 co-amoxiclav),以克服產酶菌株的抗藥性。
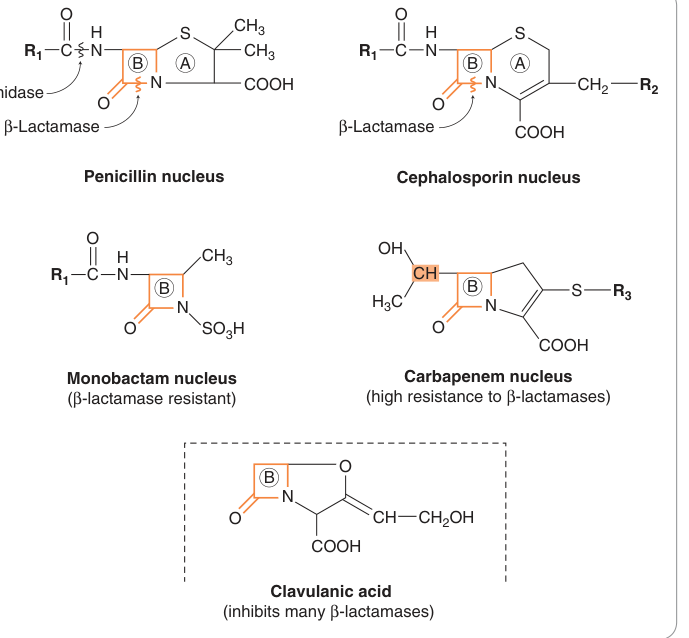
Figure 50.3:四類 β-內醯胺類抗生素與 clavulanic acid 的基本結構,標示 β-lactam 環(B)及細菌酶使抗生素失活的作用位點
藥動學
- 口服吸收因藥物而異;苄基青黴素需注射,部分可口服
- 廣泛分布於體液,但脂溶性低,不進入細胞,腦膜發炎時才可穿越血腦屏障進入 CSF
- 主要由腎臟(腎小管分泌,90%)快速排泄,半衰期短
不良反應
- 主要為過敏反應(hypersensitivity):皮疹、血清病樣反應;嚴重時可致急性過敏性休克(anaphylactic shock),雖罕見但可致死
- 口服廣效型青黴素可改變腸道菌群,引起消化道不適或繼發感染(如艱難梭菌性假膜性結腸炎)
頭孢菌素類與頭黴素類(Cephalosporins and Cephamycins)#
與青黴素作用機制相同,為廣效半合成抗生素。經歷多代演進:
- 第一代(如 cefalexin、cefradine):已大多被取代
- 第二代(如 cefuroxime、cefaclor):口服或注射
- 第三代(如 cefotaxime、ceftriaxone、ceftazidime):更廣效,部分可穿透血腦屏障
藥動學:部分可口服,多數須注射;cefotaxime、ceftriaxone 可進入 CSF;主要由腎排泄(ceftriaxone 40% 經膽汁排泄)。
不良反應
- 與青黴素有交叉過敏(約 10% 青黴素過敏者對頭孢菌素也過敏)
- 腎毒性(尤其 cefradine)、藥物性酒精不耐受、腹瀉(可因艱難梭菌引起)
臨床用途:敗血症、肺炎、腦膜炎、膽道與泌尿道感染等。
碳青黴烯類(Carbapenems)#
代表藥:亞胺培南(imipenem)、美羅培南(meropenem)、厄他培南(ertapenem)。
- 廣效,涵蓋多數需氧與厭氧革蘭氏陽性及陰性菌
- 亞胺培南需與**西司他丁(cilastatin)**合用,防止腎酶降解
- 不良反應與其他 β-內醯胺類相似;高血漿濃度可產生神經毒性
單環 β-內醯胺類(Monobactams)#
代表藥:氨曲南(aztreonam)。
- 耐大多數 β-內醯胺酶
- 僅對革蘭氏陰性需氧桿菌(如假單胞菌、腦膜炎奈瑟球菌、流感嗜血桿菌)有效
- 不與青黴素發生免疫交叉反應,適用於青黴素過敏者
糖肽類(Glycopeptides)#
萬古黴素(vancomycin)與替考拉寧(teicoplanin)。
- 抑制細菌細胞壁合成,殺菌性
- 主要對革蘭氏陽性菌有效,包括 MRSA(耐甲氧西林金黃色葡萄球菌)
- 口服用於艱難梭菌性假膜性結腸炎;靜脈注射用於多重抗藥菌感染
- 不良反應:耳毒性(ototoxicity)、腎毒性(nephrotoxicity)、注射部位靜脈炎
**達托黴素(daptomycin)**是新型脂肽類抗菌藥,抗菌譜與萬古黴素相近,常與其他藥物合用治療 MRSA。
影響蛋白質合成的抗微生物藥#
四環素類(Tetracyclines)#
代表藥:四環素(tetracycline)、多西環素(doxycycline)、米諾環素(minocycline)、替加環素(tigecycline)。
作用機制:主動轉運進入細菌後抑制蛋白質合成,屬抑菌性。
藥動學
- 口服吸收(多西環素與米諾環素幾乎完全吸收)
- 因與金屬離子(Ca²⁺、Mg²⁺、Fe²⁺、Al³⁺)螯合,牛奶、制酸劑、鐵劑會干擾吸收
- 廣泛分布於組織,可進入胎盤
不良反應
- 消化道不適(最常見)
- 與鈣結合沉積於生長中的骨骼與牙齒,引起染色、牙釉質發育不全或骨骼畸形
四環素類禁用於兒童、孕婦及哺乳期婦女。高劑量可引起肝毒性,光毒性(phototoxicity)也可能發生。
臨床用途:立克次體病、衣原體感染、布魯氏菌病(brucellosis)、萊姆病(Lyme disease)、痤瘡、呼吸道感染等。
氯黴素(Chloramphenicol)#
結合細菌核糖體 50S 亞基,廣效抑菌藥(對 H. influenzae 則有殺菌作用)。口服吸收快而完全,可進入 CSF。
最重要的不良反應是嚴重的特異性骨髓抑制(bone marrow depression),引起全血細胞減少症(pancytopenia),雖罕見但即使低劑量亦可發生。新生兒因代謝能力不足可出現「灰嬰症候群(grey baby syndrome)」,死亡率達 40%。
臨床上應保留用於其他藥物無法使用的嚴重感染(如耐藥 H. influenzae 腦膜炎)及細菌性結膜炎(局部用藥)。
胺基糖苷類(Aminoglycosides)#
代表藥:慶大黴素(gentamicin)、阿米卡星(amikacin)、鏈黴素(streptomycin)、妥布黴素(tobramycin)、新黴素(neomycin)。
作用機制:阻斷細菌核糖體起始(initiation),屬殺菌性;需氧依賴型主動轉運穿越細胞膜,對厭氧菌效果極差。
藥動學
- 高極性多陽離子,口服不吸收,通常肌肉或靜脈注射
- 不穿透血腦屏障,半衰期 2–3 小時
- 主要由腎小球過濾排泄,腎功能不全時迅速蓄積
不良反應
- 耳毒性(ototoxicity):損傷耳蝸與前庭感覺細胞,通常不可逆,可致聽力喪失或失衡;鏈黴素與慶大黴素較影響前庭,新黴素與阿米卡星較影響聽覺
- 腎毒性(nephrotoxicity):損傷腎小管,停藥後可恢復;與腎毒性藥物合用風險增加
- 神經肌肉阻斷(罕見):抑制 Ca²⁺ 依賴性乙醯膽鹼釋放
治療期間需定期監測血漿濃度,按腎功能調整劑量。與環形利尿藥合用會增強耳毒性。
大環內酯類(Macrolides)#
代表藥:紅黴素(erythromycin)、克拉黴素(clarithromycin)、阿奇黴素(azithromycin)。
作用機制:結合細菌 50S 核糖體亞基,抑制轉位(translocation)而干擾蛋白質合成;與氯黴素及克林達黴素競爭相同結合位點。
抗菌譜:類似青黴素(革蘭氏陽性菌),是青黴素過敏患者的重要替代選擇;對肺炎支原體(Mycoplasma pneumoniae)、嗜肺軍團菌(Legionella)、衣原體(Chlamydia)等有效。克拉黴素另可用於 H. pylori 除菌及鳥型分枝桿菌感染;阿奇黴素對 H. influenzae 效果更強,半衰期為紅黴素的 8–16 倍,並可大量濃縮於吞噬細胞中(溶酶體濃度可達血漿的 40 倍)。
藥動學:口服吸收良好;主要經膽汁排泄;可抑制 P450 酶系(CYP),與茶鹼等藥物有重要交互作用。
不良反應:消化道不適(最常見);紅黴素長期使用可引起膽汁淤積性黃疸(cholestatic jaundice)。
影響拓撲異構酶的抗微生物藥#
喹諾酮類(Quinolones)#
代表藥:環丙沙星(ciprofloxacin)、左氧氟沙星(levofloxacin)、莫西沙星(moxifloxacin)、諾氟沙星(norfloxacin)、氧氟沙星(ofloxacin);窄效型:萘啶酸(nalidixic acid)用於尿路感染。
作用機制:抑制拓撲異構酶 II(topoisomerase II,即 DNA 旋轉酶/DNA gyrase),阻礙 DNA 超螺旋形成,進而抑制 DNA 轉錄與複製。
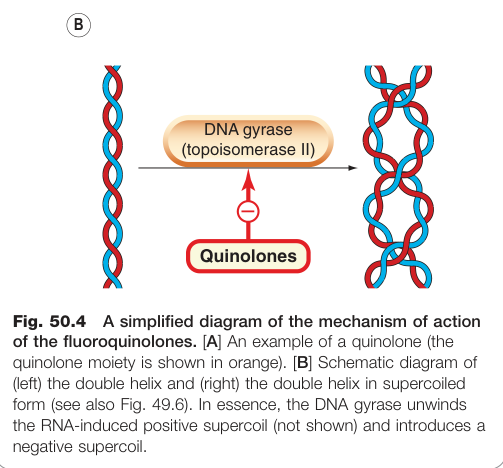
Figure 50.4:氟喹諾酮類作用機制示意,顯示藥物抑制 DNA gyrase 阻礙 DNA 超螺旋形成
抗菌譜與臨床用途
環丙沙星為最廣泛使用的氟喹諾酮類(fluoroquinolones),廣效,對腸道革蘭氏陰性桿菌、對 β-內醯胺類與胺基糖苷類耐藥菌株尤為有效;亦可對抗 P. aeruginosa、H. influenzae、N. gonorrhoeae(產青黴素酶株)等。
臨床用途
- 複雜性泌尿道感染
- 囊狀纖維化患者的 P. aeruginosa 呼吸道感染
- 慢性骨髓炎
- 傷寒菌帶原者的清除
- 淋病、攝護腺炎、炭疽
藥動學:口服吸收良好;大量分布於腎、前列腺、肺;大多不穿越血腦屏障(氧氟沙星除外);部分經 P450 代謝(可引起交互作用)。
不良反應
- 消化道不適、皮疹
- 中樞神經症狀(頭痛、暈眩);與茶鹼或非類固醇消炎藥合用時可誘發痙攣
- 年輕人可見關節病變(arthropathy)
- 醫院內艱難梭菌感染風險
環丙沙星與茶鹼有重要交互作用(抑制 P450),可導致茶鹼毒性。MRSA 感染應避免使用環丙沙星。
雜項及其他抗菌藥#
硝基咪唑類:甲硝唑(Metronidazole)#
原為抗原蟲藥,也有效對抗厭氧菌(類桿菌、梭菌、部分鏈球菌)。用於假膜性結腸炎及嚴重厭氧性感染。具雙硫侖樣作用(disulfiram-like effect),服藥期間需禁酒。
鏈陽菌素類(Streptogramins):奎奴普丁 / 達福普汀(Quinupristin / Dalfopristin)#
兩者單獨使用僅有微弱抑菌活性,合用後對多種革蘭氏陽性菌(包括 MRSA 及萬古黴素抗性腸球菌)具殺菌效果,需靜脈輸注。不良反應包括注射部位疼痛、關節痛、肌痛及消化道症狀。
林可醯胺類(Lincosamides):克林達黴素(Clindamycin)#
與大環內酯類及氯黴素作用於同一核糖體位點;對革蘭氏陽性球菌及厭氧菌(如類桿菌)有效,用於骨關節感染與葡萄球菌結膜炎。
可引發致命性假膜性結腸炎(pseudomembranous colitis),由艱難梭菌(Clostridium difficile)毒素引起,需立即停藥並以甲硝唑或萬古黴素治療。
噁唑烷酮類(Oxazolidinones):利奈唑胺(Linezolid)#
抑制 N-甲醯甲硫胺醯-tRNA 與 70S 核糖體的結合;對 MRSA、耐青黴素肺炎鏈球菌(penicillin-resistant Streptococcus pneumoniae)及萬古黴素抗性腸球菌(VRE)有效。口服或靜脈均可使用,為多重抗藥嚴重感染的保留用藥。不良反應:血小板減少症(thrombocytopenia)、腹瀉;因具單胺氧化酶抑制作用,需注意相關交互作用。
夫西地酸(Fusidic Acid)#
窄效固醇類抗生素,主要對革蘭氏陽性菌有效,抑制細菌蛋白質合成;可滲入骨骼,用於葡萄球菌性骨感染(常與其他抗葡萄球菌藥合用)。單獨使用全身性療程易出現抗藥性。
硝基呋喃類(Nitrofurantoin)#
- 對多種革蘭氏陽性及陰性菌有效,抗藥性少見
- 口服快速完全吸收,迅速由腎排泄
- 僅用於泌尿道感染(urinary tract infections,UTIs)
- 不良反應:消化道不適、過敏反應、肝毒性、周邊神經病變
多黏菌素類(Polymixins):多黏菌素 B(polymixin B)、黏菌素(colistin)#
陽離子清潔劑,破壞革蘭氏陰性菌外膜;對假單胞菌及大腸桿菌等迅速殺菌。因神經毒性與腎毒性顯著,僅用於腸道滅菌及局部治療(耳、眼、皮膚感染)。
抗分枝桿菌藥(Antimycobacterial Agents)#
結核病(Tuberculosis)的治療#
結核病(tuberculosis)由結核分枝桿菌(Mycobacterium tuberculosis)引起,分枝桿菌可在巨噬細胞(macrophages)中存活,需 Th1 細胞激活後才能被消滅。目前全球約有 20 億人感染,每年新增逾 900 萬病例,死亡約 180 萬人,多重抗藥性(multidrug resistance,MDR)問題嚴峻。
為預防抗藥性,採複合藥物療法:
- 初始期(約 2 個月):異菸鹼醯胺(isoniazid)+ 利福平(rifampicin)+ 吡嗪醯胺(pyrazinamide)± 乙胺丁醇(ethambutol)
- 後續期(約 4 個月):異菸鹼醯胺 + 利福平
第一線藥物#
異菸鹼醯胺(Isoniazid)
- 前驅藥,由細菌酶活化後抑制分枝菌酸(mycolic acids)的合成
- 對靜止中細菌具抑菌性,對分裂中細菌具殺菌性;可進入細胞內及乾酪樣(caseous)病灶
- 代謝依賴乙醯化(acetylation),由遺傳決定「慢乙醯化者(slow acetylator)」或「快乙醯化者」,慢乙醯化者治療效果較佳
- 不良反應:皮疹、肝毒性、血液病變;最重要的是吡哆醇(pyridoxine/維生素 B6)缺乏引起的周邊神經病變,在營養不良患者中尤為常見;另可抑制苯妥英、乙琥胺、卡馬西平的代謝而增強其毒性
利福平(Rifampicin,rifampin)
- 結合並抑制細菌 DNA 依賴型 RNA 聚合酶(DNA-dependent RNA polymerase)
- 口服活性高,可進入 CSF、吞噬細胞,並殺滅細胞內結核桿菌
- 抗藥性可迅速在單步驟中發展(染色體突變)
- 可將唾液、痰液、淚液、汗液染成橘色
- 不良反應:皮疹、消化道不適、偶見肝毒性;誘導肝臟代謝酶(CYP 誘導),使華法林、口服避孕藥、糖皮質激素等多種藥物代謝加快,藥效降低
乙胺丁醇(Ethambutol)
- 機制未明,抑菌性;進入細菌後需 24 小時才顯效
- 最重要不良反應:視神經炎(optic neuritis),與劑量相關,初期表現為紅綠色盲,進而視力下降;治療期間需監測色覺
吡嗪醯胺(Pyrazinamide)
- 中性 pH 下無活性,在巨噬細胞吞噬溶酶體(phagolysosome)的酸性環境中有效,為殺滅細胞內分枝桿菌的關鍵
- 不良反應:尿酸升高(可引發痛風)、消化道不適、肝毒性(高劑量)
第二線藥物#
- 卷曲黴素(capreomycin):肌肉注射,不良反應含腎毒性與第八對腦神經損傷
- 環絲胺酸(cycloserine):廣效型,抑制肽聚糖合成;口服,可穿透 CSF;主要不良反應為中樞神經系統毒性
- 鏈黴素(streptomycin):胺基糖苷類,肌肉注射;耳毒性(主要影響前庭)與腎毒性
麻瘋病(Leprosy)的治療#
麻瘋病由麻瘋分枝桿菌(Mycobacterium leprae)引起,自 1982 年世界衛生組織推行多重藥物療法(multidrug therapy)後,全球流行率已下降 90%,目前每年約新增 20 萬病例(集中於印度次大陸、巴西及莫三比克)。
- 少桿菌型(paucibacillary/tuberculoid)麻瘋:氨苯碸(dapsone)+ 利福平,療程 6 個月
- 多桿菌型(multibacillary/lepromatous)麻瘋:氨苯碸 + 利福平 + 氯苯吩嗪(clofazimine),療程至少 2 年
氨苯碸(Dapsone)
- 化學上與磺胺類相似,可能透過抑制葉酸合成發揮作用(PABA 可拮抗之)
- 口服吸收良好,廣泛分布;半衰期 24–48 小時
- 不良反應:溶血性貧血(通常輕度)、高鐵血紅蛋白血症、過敏性皮炎、發燒、神經病變;葡萄糖-6-磷酸脫氫酶(G6PD)缺乏者風險更高
氯苯吩嗪(Clofazimine)
- 複雜結構染料,機制可能涉及 DNA;另具抗發炎活性
- 口服後在單核吞噬系統蓄積,半衰期長達 8 週,抗痲瘋效果延遲 6–7 週才顯現
- 不良反應:皮膚與尿液呈紅色、皮損紫黑色,及消化道不適
新型抗菌藥物的研發#
自 1950–1980 年代「英雄時代」之後,新抗生素的發現急劇減少,近數十年僅有極少數全新類別藥物問世,但細菌的多重抗藥性問題日益嚴峻。目前的研究方向包括:
- 天然來源(植物、細菌)的新型化合物篩選
- 生物資訊學(bioinformatics)結合病原基因組測序,鎖定新靶點
- 細菌毒力因子(virulence factors)的針對性研究
- 新型藥效動力學(pharmacodynamic)分析方法