概覽#
動作電位(action potential)的產生依賴電壓閘控鈉離子通道(voltage-gated sodium channel),膜電位去極化時通道短暫開啟,驅動神經與肌肉的電興奮性(electrical excitability)。改變鈉離子通道功能的方式主要有兩類:
- 阻斷通道:降低興奮性
- 修改閘控行為(gating):依據效果可增加或抑制興奮性
局部麻醉劑(local anaesthetic)是臨床上透過阻斷鈉離子通道來消除局部神經傳導的主要藥物;其他具有相同作用的還包括部分抗癲癇藥、止痛藥及第 I 類抗心律不整藥。
局部麻醉劑#
歷史#
南美洲原住民咀嚼古柯葉(coca leaf)已有數千年歷史,知曉其對口腔與舌頭有麻木效果。1860 年可卡因(cocaine)被分離出來,後於 1884 年由眼科醫師 Carl Köller 報告可在眼部手術中產生可逆性角膜麻醉,隨即被引入牙科與一般外科。1905 年普魯卡因(procaine)問世,此後多種合成替代品相繼開發。
化學結構#
- 局部麻醉劑分子由**芳香環(aromatic group)經酯鍵或醯胺鍵(ester or amide bond)連接鹼性胺基側鏈(basic amine side-chain)**所構成
- 為弱鹼,pKa 值多在 8–9 之間,生理 pH 下主要以離子化形式存在
- 酯類(如 procaine、tetracaine):被血漿與組織的非特異性酯酶迅速水解,半衰期短
- 醯胺類(如 lidocaine、bupivacaine):主要在肝臟代謝,半衰期較長(約 1–2 小時)
季銨衍生物(quaternary derivative)如 QX-314,無論 pH 均完全離子化,無法穿透神經鞘而不具臨床局麻效果,但為重要的實驗工具。Benzocaine 無鹼性基團,屬非典型局麻藥。
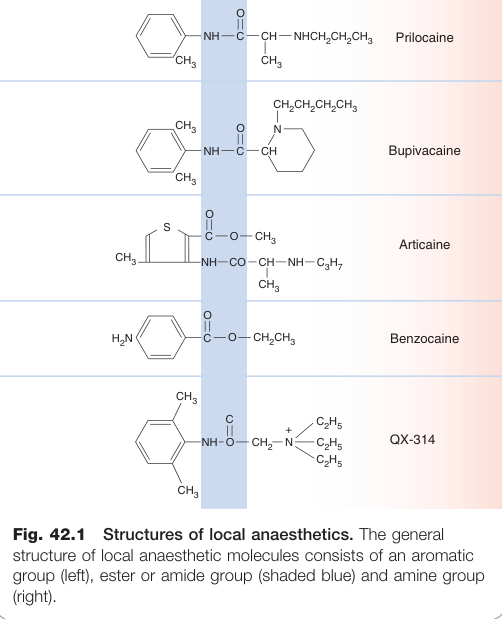
Figure 42.1:局部麻醉劑的化學結構,由芳香環(左)、酯或醯胺連接基(藍色區)及鹼性胺基側鏈(右)三部分組成
作用機制#
局部麻醉劑藉由物理性阻塞跨膜孔道,與通道蛋白 S6 跨膜螺旋域上的胺基酸殘基結合,阻止電壓依賴性 Na⁺ 電導上升,從而抑制動作電位的起始與傳導。
pH 依賴性
- 鹼性環境(離子化比例低)活性增強;酸性環境活性降低
- 藥物須以非離子化形式穿透神經鞘與軸突膜,到達通道內側的結合位點後,**離子化形式(BH⁺)**為主要阻斷分子
- 臨床意義:發炎組織偏酸,局麻效果往往較差
使用依賴性阻斷(use-dependent block)
- 通道開啟次數越多,阻斷程度越深
- 原因一:阻斷分子在通道開啟時更容易進入
- 原因二:許多局麻劑對**失活態(inactivated state)**的親和力高於靜息態
使用依賴性阻斷是第 I 類抗心律不整藥與抗癲癇藥重要的藥理特性,在高頻動作電位發放時尤為明顯。
通道狀態與藥物進入途徑
| 途徑 | 形式 | 特性 |
|---|---|---|
| 親水途徑(hydrophilic pathway) | BH⁺(離子化) | 從通道內側開口進入,具使用依賴性 |
| 親脂途徑(hydrophobic pathway) | B(非離子化) | 直接從膜相進入,不具使用依賴性 |
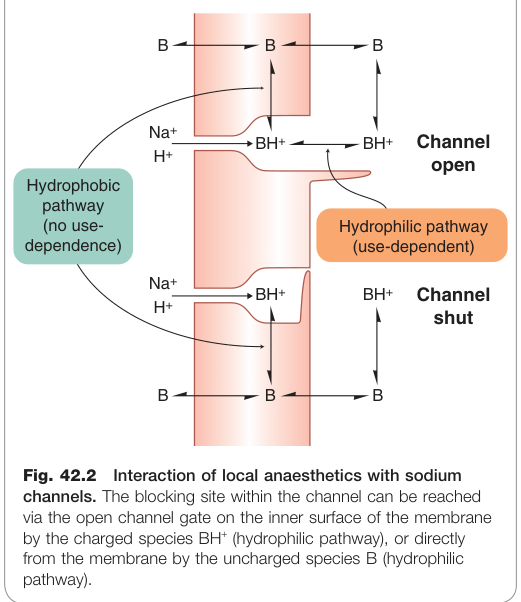
Figure 42.2:局部麻醉劑與鈉離子通道的交互作用,顯示帶電分子經開放通道閘進入、中性分子經疏水膜路徑進入的兩種阻斷途徑
神經纖維選擇性
- 細徑纖維比粗徑纖維更容易被阻斷
- Aδ 與 C 纖維(傷害性感覺)最先被阻斷,觸覺、本體感覺及運動軸突相對耐受
- 儘管如此,實際上無法只阻斷疼痛而完全保留其他感覺
低濃度靜脈注射 lidocaine(lignocaine)可抑制神經病變疼痛(neuropathic pain)中感覺神經元的自發性放電。
常用藥物比較#
| 藥物 | 起效 | 持續時間 | 特點 |
|---|---|---|---|
| Lidocaine(lignocaine) | 快 | 中等 | 臨床最廣泛使用;靜脈注射可治療心室性心律不整 |
| Bupivacaine | 慢 | 長 | 廣泛用於硬脊膜外與脊椎麻醉;心毒性較高 |
| Levobupivacaine | 慢 | 長 | Bupivacaine R(−) 異構體;心毒性與 CNS 抑制較低 |
| Ropivacaine | 慢 | 長 | 類似 bupivacaine,心毒性較低 |
| Prilocaine | 中 | 中等 | 無血管擴張;大劑量可導致高鐵血紅蛋白血症(methaemoglobinaemia) |
| Tetracaine(amethocaine) | 極慢 | 長 | 主要用於脊椎與角膜麻醉 |
| Articaine | 快 | 短 | 用於牙科;少數患者可出現藥物消失後仍持續的麻木感 |
| Cocaine | 中 | 中等 | 唯一具擬交感作用的局麻藥(抑制去甲腎上腺素再攝取);限用於上呼吸道黏膜 |
不良反應#
中樞神經系統(CNS)
- 低濃度:抑制效果為主(鎮靜)
- 高濃度:轉為興奮——躁動、震顫、癲癇發作
- 更高劑量:深度 CNS 抑制、呼吸停止
- 例外:cocaine 在相對低劑量即產生欣快感(euphoria),機制在於抑制單胺再攝取
心血管系統
- 心肌抑制:抑制心肌 Na⁺ 電流 → [Na⁺]ᵢ 下降 → 細胞內 Ca²⁺ 儲存減少 → 收縮力下降
- 傳導阻斷:可致房室傳導阻斷或心律不整
- 血管擴張:動脈小血管直接舒張+交感神經阻斷 → 血壓急速下降,可危及生命
- Cocaine 例外:因抑制去甲腎上腺素再攝取,反而導致心跳加速、心輸出量增加、血壓上升
其他
- 過敏反應:通常為接觸性皮炎,偶見急性過敏反應
- 高鐵血紅蛋白血症:prilocaine 大劑量時,因毒性代謝物所致
- 黏膜刺激:cocaine
Bupivacaine 的心毒性高於其他局麻藥,意外靜脈注射時風險尤大。建議使用 levobupivacaine 或 ropivacaine 以降低心毒性風險。
藥物動力學#
- 酯類(tetracaine 等):血漿膽鹼酯酶快速水解,半衰期短;procaine 水解為對胺基苯甲酸(p-aminobenzoic acid),會干擾磺胺類藥物的抗菌效果
- 醯胺類(lidocaine、prilocaine 等):主要在肝臟進行 N-去烷基化,代謝物常具藥理活性
- Benzocaine:水溶性極低,用於乾粉敷料或含片,緩慢釋放產生持久的表面麻醉
多數局部麻醉劑具有直接血管擴張作用,加速全身吸收。因此常加入腎上腺素(adrenaline / epinephrine)或felypressin(短效血管加壓素類似物)作為血管收縮劑,以延長局部作用並降低全身毒性。
加入腎上腺素的局麻劑不可用於手指或腳趾,以免引起缺血性組織損傷。
臨床給藥途徑#
| 方式 | 應用 | 注意事項 |
|---|---|---|
| 表面麻醉 | 鼻腔、口腔、支氣管、角膜、尿路 | 對皮膚效果差(EMLA 乳膏除外) |
| 浸潤麻醉 | 局部手術直接注射至組織 | 範圍不宜過大,避免全身毒性 |
| 靜脈區域麻醉 | 肢體手術(止血帶充氣後注射) | 止血帶至少充氣 20 分鐘再放鬆 |
| 神經阻斷 | 臂叢、肋間、牙科神經 | 精確定位重要,起效可能較慢 |
| 脊椎麻醉 | 腹部、骨盆、下肢手術 | 風險:心搏過緩、低血壓、尿瀦留 |
| 硬脊膜外麻醉 | 手術、無痛分娩 | 與鴉片類藥物合用可加強鎮痛效果 |
其他影響鈉離子通道的藥物#
河魨毒素(Tetrodotoxin, TTX)與石房蛤毒素(Saxitoxin, STX)#
- TTX:由海洋細菌產生,蓄積於河魨(puffer fish)組織;日本視河魨為珍饈,但處理不當仍可致命
- STX:由「赤潮」中大量增殖的海洋微生物產生,可在貝類中蓄積
兩者共同特點:
- 從膜外側作用於鈉離子通道,與傳統局麻藥不同
- 分子帶正電的胍基(guanidinium moiety)嵌入通道孔口,阻塞其外側入口
- 無閘控依賴性:結合與解離不受通道開關狀態影響(無使用依賴性)
- 部分鈉離子通道亞型(如心肌通道及神經病變疼痛中上調的通道)對 TTX 不敏感
TTX 與 STX 因取得困難且組織穿透性差(親水性高),臨床不適合作為局麻藥使用,但為研究鈉離子通道分離與選殖的重要實驗工具。
影響鈉離子通道閘控的藥物#
某些物質以促進通道開啟的方式修改閘控行為,包括:
- 毒素:蟾蜍皮膚毒素 batrachotoxin、蠍毒、海葵毒素
- 植物生物鹼:veratridine
- 殺蟲劑:DDT、除蟲菊酯(pyrethrins)
效果:
- 降低通道活化閾值(在更負的膜電位即可開啟)
- 抑制通道失活,使去極化後通道無法關閉
- 導致膜超興奮性 → 自發放電 → 最終持續去極化而失去興奮性
這類物質影響心臟(引起期外收縮、心室顫動)、神經與肌肉(抽搐、癲癇)。DDT 等高親脂性殺蟲劑可穿透昆蟲體壁而有效。此類藥物目前僅作為實驗工具,無臨床用途。