概述#
成人中樞神經系統(CNS)的死亡神經元無法再生,軸突被切斷後末梢也無法重建。因此,任何造成神經元死亡的病理過程,後果幾乎都是不可逆的。神經退化性疾病(neurodegenerative diseases)目前的治療相當有限,僅帕金森氏症(Parkinson’s disease, PD)有較成熟的藥物可用。
本章涵蓋三大主題:
- 阿茲海默症(Alzheimer’s disease, AD):最常見的慢性神經退化疾病
- 帕金森氏症(Parkinson’s disease, PD):以多巴胺缺乏為核心的運動障礙
- 缺血性腦損傷(ischaemic brain damage)/ 中風(stroke):急性腦缺血,致病機轉與慢性退化疾病不同
蛋白質錯誤折疊與聚集#
許多慢性神經退化疾病的共同病因,是正常蛋白質發生**錯誤折疊(misfolding)**後聚集。
- 錯誤折疊使蛋白質的疏水性殘基暴露在表面,促進聚集與膜結合
- 聚集過程:單體 → 可溶性寡聚體(oligomer)→ 不可溶的大型聚集體(insoluble aggregate)
- 這些聚集體可形成稱為**澱粉樣沉積(amyloid deposits)**的顯微結構,為神經退化疾病的病理特徵
- 腦部具有保護機制,包括**伴侶蛋白(chaperone proteins)和泛素化(ubiquitination)**降解系統;若這些機制不堪負荷,沉積便會累積
以下疾病均與蛋白質錯誤折疊有關:阿茲海默症(Aβ、Tau)、帕金森氏症(α-突觸核蛋白,α-synuclein)、庫賈氏症(Creutzfeldt-Jakob disease, CJD)(朊蛋白,prion protein)、亨廷頓氏症(Huntington’s disease)(huntingtin)、肌萎縮性脊髓側索硬化症(amyotrophic lateral sclerosis, ALS)(超氧化物歧化酶,superoxide dismutase)。
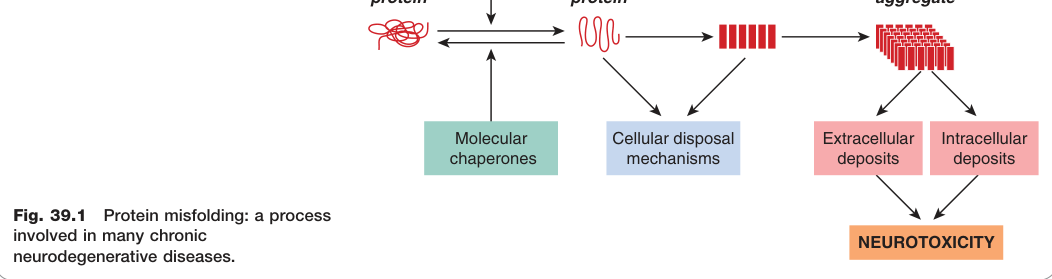
Figure 39.1:多種慢性神經退化疾病共有的蛋白質錯誤折疊病理過程
神經元死亡機轉#
壞死與凋亡#
- 壞死(necrosis):急性損傷引起,細胞膨脹、溶解,釋放內容物引發炎症;鈣離子超載是核心事件
- 凋亡(apoptosis):程序性細胞死亡,細胞系統性解體後由巨噬細胞清除,不引發炎症;在慢性(AD、PD)和急性(中風)神經退化中均可發生
- 兩者並非截然二分:輕度興奮性毒性或氧化壓力可誘發凋亡,重度則直接造成壞死
神經元存活依賴神經生長因子(nerve growth factor)和腦源性神經營養因子(brain-derived neurotrophic factor),這些因子透過 Bcl-2/Bax 比例調節凋亡。
興奮性毒性(Excitotoxicity)#
麩胺酸(glutamate)過量是神經元死亡的重要途徑:
- **NMDA、AMPA 及代謝型受體(metabotropic receptor)**活化 → 細胞去極化 → 解除 Mg²⁺ 對 NMDA 通道的阻塞 → Ca²⁺ 大量湧入
- 電壓門控鈣離子通道(VDCC)開放,進一步增加 Ca²⁺ 內流
- 去極化抑制麩胺酸再回收,胞外麩胺酸濃度持續上升(正回饋)
- 細胞內 Ca²⁺ 超載(Ca²⁺ overload)→ 活化蛋白酶(calpains)、脂酶、一氧化氮合酶(NOS)→ 產生活性氧物種(reactive oxygen species, ROS)、膜損傷、DNA 斷裂
腦缺血時,大量麩胺酸釋放觸發典型的興奮性毒性級聯反應,此為中風神經損傷的核心機制。
針對興奮性毒性的藥物嘗試(動物實驗有效,臨床均告失敗):
- 麩胺酸受體拮抗劑(NMDA 拮抗劑:memantine 對中重度 AD 有輕微效益)
- 鈣離子通道阻斷劑
- 自由基清除劑
- Riluzole:同時抑制麩胺酸釋放和突觸後作用,可輕微延緩 ALS 惡化
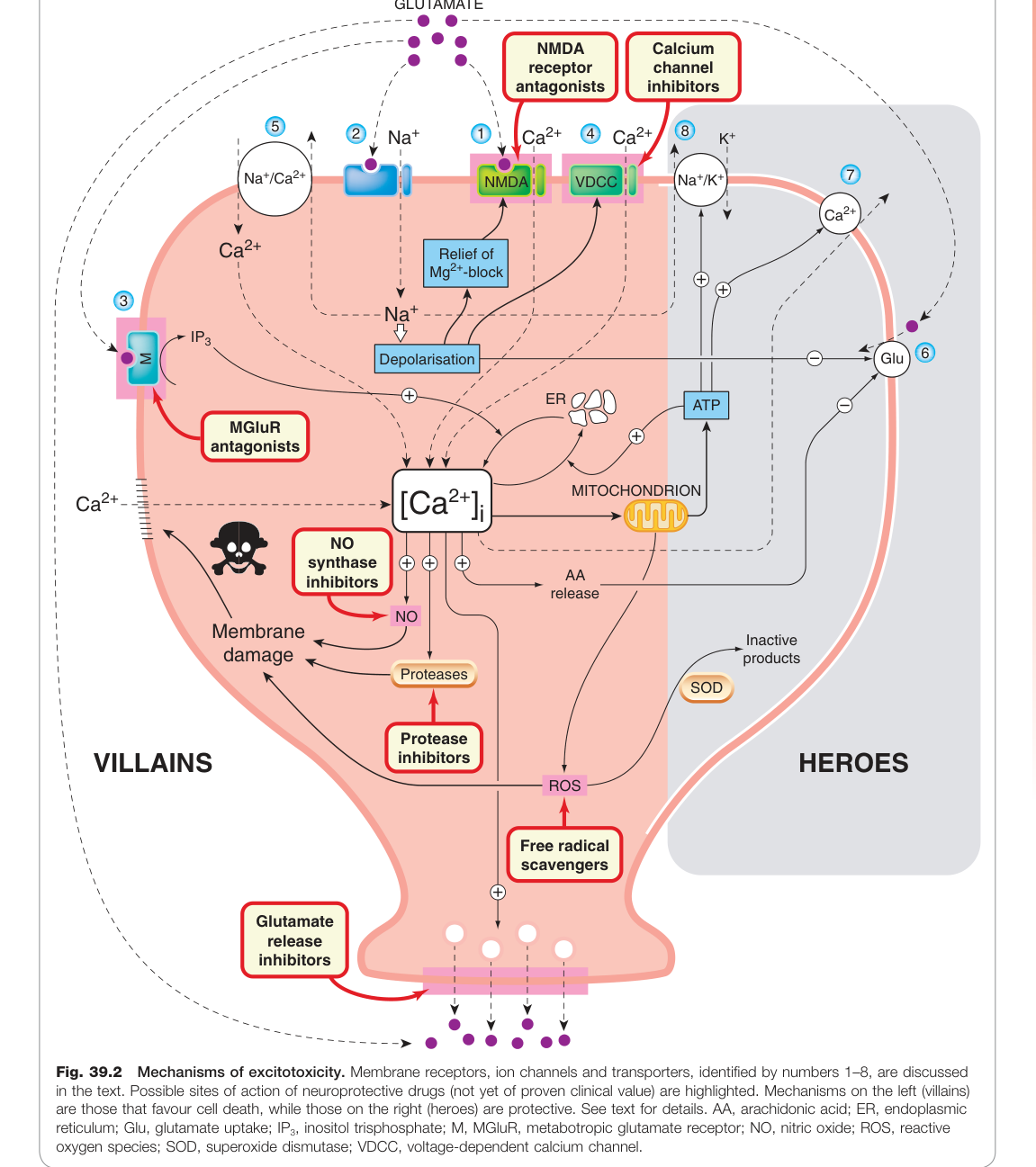
Figure 39.2:興奮性毒性的機轉與潛在神經保護藥物的作用位點
氧化壓力(Oxidative stress)#
腦的高能量需求幾乎完全依賴粒線體氧化磷酸化,此過程可產生 ROS 副產物:
- 正常情況下由超氧化物歧化酶(SOD)、過氧化氫酶(catalase)及抗氧化劑(維生素 C、維生素 E、麩胱甘肽)維持平衡
- 粒線體功能障礙是多種神經退化疾病的共同特徵,粒線體損傷同時引發氧化壓力和凋亡(釋放細胞色素 c)
- ALS 與 SOD 基因突變有關;粒線體複合體酶突變則可能導致遺傳性或年齡相關的神經退化
缺血性腦損傷(中風)#
中風是歐美第三大死因,70% 的非致命中風是最常見的失能原因。約 85% 為缺血性(多為腦動脈血栓),15% 為出血性。
病理生理#
- 梗塞核心(core):血流中斷後數分鐘內神經元發生不可逆壞死
- 半影區(penumbra):外圍受損區,興奮性毒性和發炎在數小時內造成進行性凋亡,是治療介入的時間窗口
- 再灌流(reperfusion)可能因產生大量 ROS 而造成二次傷害
治療#
- 阿替普酶(alteplase):重組組織型纖溶酶原活化劑(rt-PA),靜脈注射溶解血栓,需在3 小時內給予,且必須先排除出血性中風(電腦斷層掃描確認)
- 超過 37 種針對興奮性毒性的神經保護藥物已完成逾 114 項臨床試驗,全數失敗
- 預防(控制血壓、阿斯匹靈、防治動脈粥狀硬化)目前優於治療
中風治療至今仍是藥理學的未竟之業;臨床試驗困難且昂貴,動物模型預測性低,成功率為零。
阿茲海默症#
病理特徵#
AD 的盛行率隨年齡急遽上升(65 歲約 5%,95 歲約 90%),其病理特徵為:
- 細胞外澱粉樣斑塊(amyloid plaques):由 β-澱粉樣蛋白(Aβ)組成,源自澱粉樣前驅蛋白(amyloid precursor protein, APP)經 β 和 γ-分泌酶(secretase)切割而來
- 神經纖維纏結(neurofibrillary tangles):由過度磷酸化的 Tau 蛋白組成,損害軸突運輸
- 膽鹼性神經元喪失:基底前腦核(basal forebrain nuclei)的膽鹼性神經元選擇性退化,是記憶與認知障礙的主要原因
發病機轉#
- APP 處理異常:Aβ42 比 Aβ40 更具聚集傾向,是澱粉樣斑塊形成的主要元凶
- 家族性 AD(少見):與 APP 基因突變、presenilin 基因突變(提高 γ-分泌酶活性)有關,均導致 Aβ42 生成增加
- ApoE4 突變:影響 Aβ 清除,增加散發性 AD 風險
- Tau 病理:Aβ 促進 Tau 磷酸化,過度磷酸化的 Tau 又促進澱粉樣沉積,兩者具協同神經毒性
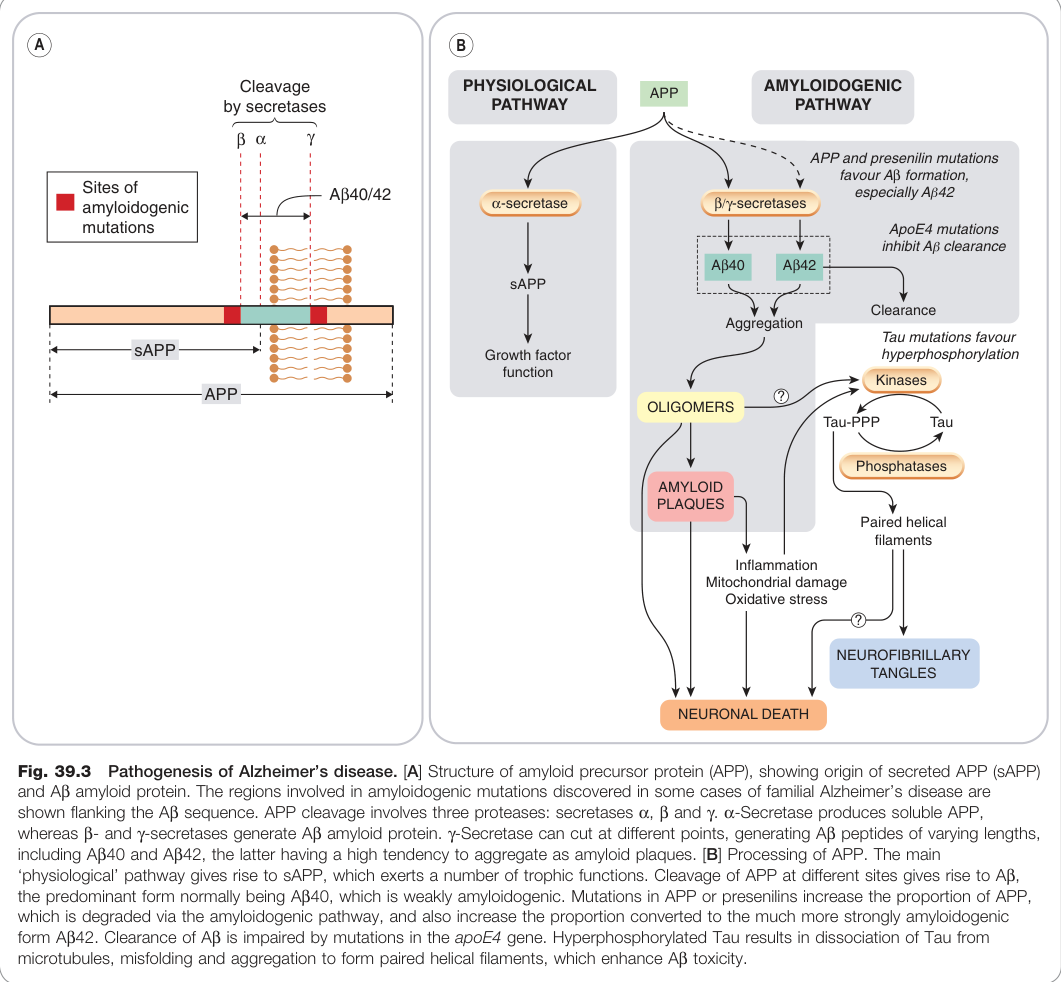
Figure 39.3:阿茲海默症病理機轉,包括 APP 結構、Aβ 來源及澱粉樣斑塊形成
唐氏症(Down’s syndrome)患者 21 號染色體多了一份,APP 基因過表達,導致早期出現類 AD 失智。
藥物治療#
膽鹼酯酶抑制劑(Cholinesterase inhibitors)#
| 藥物 | 選擇性 | 劑量頻率 | 備註 |
|---|---|---|---|
| Tacrine | AChE + BuChE,非選擇性 | 一日四次 | 已少用;肝毒性 |
| Donepezil | CNS 選擇性,AChE | 一日一次 | 耐受性最好 |
| Rivastigmine | CNS 選擇性 | 一日兩次 | 逐漸增量以減少副作用 |
| Galantamine | AChE + BuChE;另有菸鹼受體增效作用 | 一日兩次 | — |
- 對 40% AD 患者的記憶與認知測驗有輕微改善,但未能延緩疾病進程
- 常見副作用:噁心、腹痛、腹瀉等膽鹼性效應
Memantine#
- NMDA 受體弱拮抗劑,口服有效
- 對中重度 AD 有輕微認知改善,副作用少,半衰期長
- 不具神經保護作用
抑制神經退化的研究進展(尚未臨床成功)#
- β/γ-分泌酶抑制劑:可降低 Aβ 生成,但多有免疫毒性和腸道毒性
- Tau 激酶抑制劑:針對 Tau 過度磷酸化,開發中
- Aβ 免疫療法:Aβ 抗體注射可逆轉動物模型斑塊,人體試驗因神經炎症副作用叫停;單株抗體持續臨床試驗中
- 金屬螯合劑(如 clioquinol):可溶解 Aβ 斑塊,但因毒性無法廣泛使用
- NSAIDs:流行病學顯示某些 NSAIDs(如 ibuprofen)可降低 AD 發生率,但臨床試驗結果負面
帕金森氏症#
臨床特徵#
帕金森氏症是一種主要影響老年人的進行性運動障礙,四大核心症狀:
- 運動不能(hypokinesia):自發運動減少,啟動與停止均困難
- 靜止性震顫(tremor at rest):常從手部開始,自主運動時減輕
- 肌肉強直(rigidity):被動活動時阻力增加
- 認知障礙:程度不一,晚期非運動症狀(失智、憂鬱、自律神經失調)往往更為顯著
神經化學基礎#
- 1960 年,Hornykiewicz 發現 PD 患者黑質(substantia nigra)和紋狀體(corpus striatum)的多巴胺含量極低(< 正常值 10%)
- 症狀在紋狀體多巴胺降至 20-40% 以下才出現
- 紋狀體膽鹼性中間神經元活性受多巴胺抑制;多巴胺缺乏時膽鹼性活性過高,亦是症狀來源
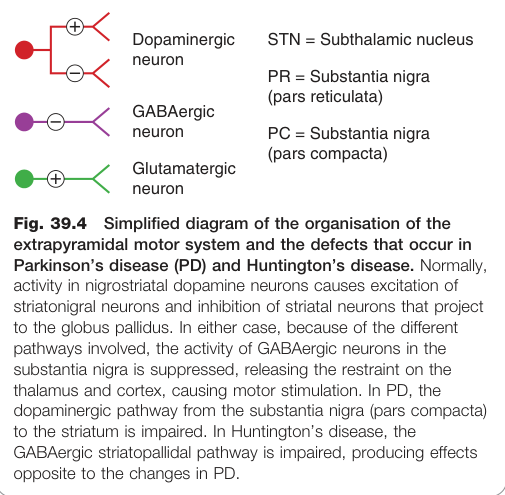
Figure 39.4:錐體外路運動系統組織及帕金森氏症與亨廷頓氏症的神經迴路缺損
發病機轉#
- Lewy 體(Lewy bodies):含大量 α-突觸核蛋白(α-synuclein)的細胞內蛋白聚集體,為 PD 病理標誌
- α-synuclein 突變(家族性 PD)或 parkin 突變(影響蛋白降解)均與 Lewy 體形成有關
- MPTP 毒素模型:1982 年加州藥物成癮者因攝入 MPTP 汙染物急速出現重度 PD;MPTP 被 MAO-B 轉化為 MPP⁺,選擇性破壞黑質紋狀體多巴胺神經元
- 粒線體功能障礙和氧化壓力是神經退化的重要共同因素
藥物治療#
左旋多巴(Levodopa)#
- 一線治療,必須與多巴脫羧酶抑制劑(DDC inhibitor)(carbidopa 或 benserazide)合用,可將所需劑量降低 10 倍,並減少周邊副作用
- 約 80% 患者初始改善明顯(強直與運動不能最有效),但療效隨疾病進展逐漸下降
- 合用 COMT 抑制劑(entacapone)可減少「劑末效應(end-of-dose)」波動
主要副作用:
- 遲發性運動障礙(dyskinesia):2 年內多數患者出現不自主扭動動作,治療窗口逐漸縮窄
- 「開-關」效應(on-off effect):突發性運動凍結,與左旋多巴血漿濃度波動有關
- 急性副作用:噁心(可用 domperidone 預防)、姿勢性低血壓、精神症狀(幻覺、混亂)
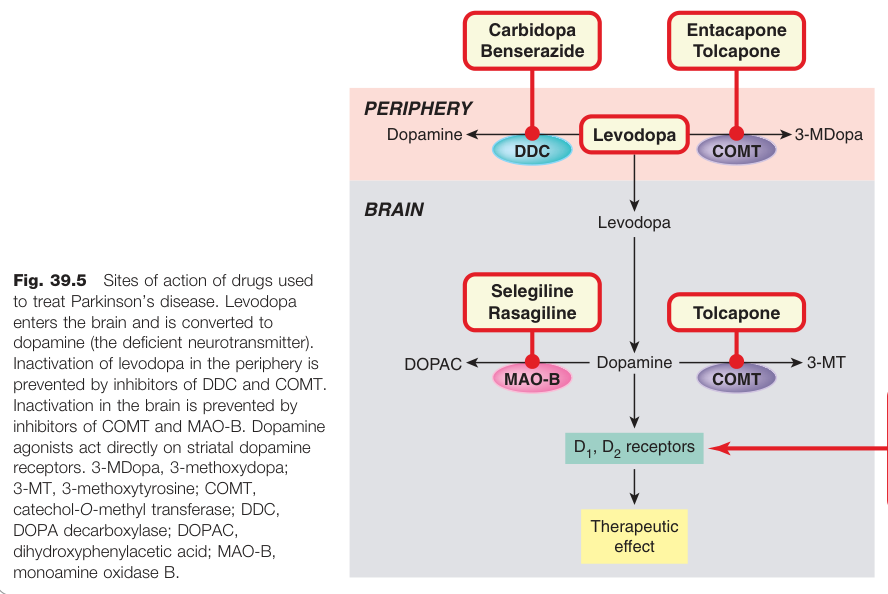
Figure 39.5:治療帕金森氏症藥物的作用位點,包括 levodopa 及各種輔助療法
多巴胺受體致效劑(Dopamine agonists)#
- Pramipexole、Ropinirole(D2/D3 選擇性,非麥角類):耐受性優於舊型麥角類藥物,但可能引起嗜睡及衝動控制障礙(病態賭博、暴食)
- Rotigotine:貼片劑型,持續給藥
- Apomorphine:注射劑型,用於「關期(off period)」急救;需合用口服止吐藥
MAO-B 抑制劑#
- Selegiline(司來吉蘭):選擇性 MAO-B 抑制劑,保護多巴胺免於降解;其預防 MPTP 神經毒性的機制也提示其可能具神經保護作用,但臨床試驗未證實此點
- Rasagiline(雷沙吉蘭):與 selegiline 相似但不產生安非他命代謝物,近期試驗顯示可能輕微延緩疾病進程
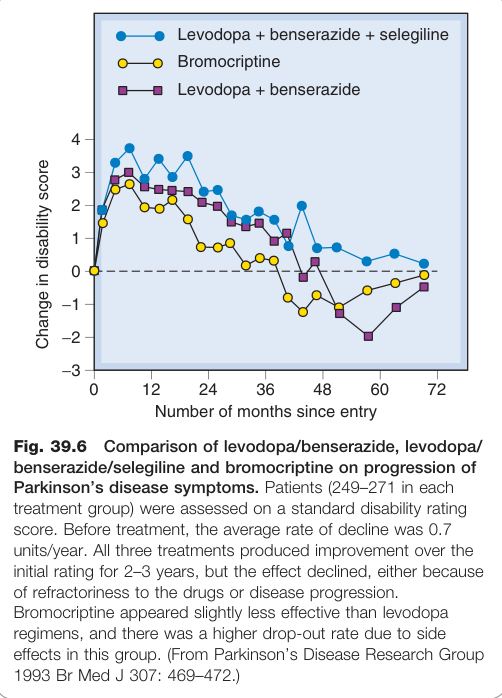
Figure 39.6:levodopa/benserazide、levodopa/benserazide/selegiline 及 bromocriptine 對帕金森氏症症狀進展的比較
其他藥物#
- Amantadine:機轉不明(可能促進多巴胺釋放),效果弱、持續時間短
- 毒蕈鹼受體拮抗劑(如 benztropine):主要用於抗精神病藥物引起的帕金森症狀
目前所有 PD 藥物均為症狀緩解,無一能阻止神經退化進程。
神經移植與腦深層電刺激#
- 將胎兒腦細胞移植入紋狀體已進行試驗,部分患者有功能改善,但少數出現嚴重遲發性運動障礙;幹細胞移植是未來方向
- **腦深層電刺激(deep brain stimulation)**刺激丘腦下核(subthalamic nucleus),可改善運動症狀,但對認知症狀無效
亨廷頓氏症#
亨廷頓氏症(Huntington’s disease, HD)是體染色體顯性遺傳的進行性神經退化疾病,屬三核苷酸重複疾病(CAG 重複),mutant huntingtin 含 40 個以上麩醯胺酸重複序列,重複數越多發病越早。
- 退化主要影響皮質和紋狀體,導致失智和嚴重的舞蹈病動作(choreiform movements)
- 紋狀體 GABA 能神經元喪失 → 多巴胺系統過度活躍(與 PD 相反)
- 治療以緩解運動症狀為主(tetrabenazine、多巴胺拮抗劑、baclofen),無法延緩病程
朊病毒(Prion)疾病#
包括人類庫賈氏症(CJD)、變異型 CJD(vCJD,與 BSE「狂牛症」有關)及動物羊搔癢症(scrapie)。
- 致病因子為異常折疊的朊蛋白(PrP^Sc),可將正常 PrP^C 招募轉化,形成鏈式反應
- PrP 基因剔除小鼠對感染具有抵抗力,證明 PrP^Sc 是致病關鍵
- 目前無有效治療;奎納克林(quinacrine)和氯丙嗪(chlorpromazine)在動物模型可抑制 PrP^Sc 聚集,仍在人體試驗階段