本章介紹中樞神經系統(central nervous system, CNS)最主要的兩類氨基酸傳遞物:興奮性的麩胺酸(glutamate)與抑制性的 γ-胺基丁酸(GABA)及甘胺酸(glycine)。
興奮性氨基酸#
麩胺酸作為 CNS 傳遞物#
- **L-麩胺酸(L-glutamate)**是 CNS 中最主要且普遍分布的興奮性傳遞物;天門冬胺酸(aspartate)在某些腦區亦扮演相似角色。
- 麩胺酸的發現歷程漫長。1950 年代 Curtis 團隊證實其興奮效應,但當時研究者難以相信如此平凡的代謝物竟是傳遞物。直至 Watkins(John Watkins)在 Bristol 發現興奮性氨基酸(excitatory amino acid, EAA)拮抗劑,才確立麩胺酸的生理角色。
- EAA 廣泛參與腦內神經傳遞,因此藥物作用範圍難以限縮,導致治療效益與副作用並存。
代謝與釋放#
- 麩胺酸在 CNS 中濃度遠高於其他組織,主要來源是葡萄糖(經 Krebs 循環)或膠細胞合成的麩醯胺(glutamine),後者被神經元攝取後轉回麩胺酸。
- 麩胺酸與抑制性氨基酸(GABA、甘胺酸)共用合成路徑(圖 37.1),因此干擾任一步驟都會同時影響興奮性與抑制性傳遞。
- 麩胺酸儲存於突觸小泡,以 Ca²⁺ 依賴性胞吐方式釋放。
- 釋放後的麩胺酸主要由**興奮性氨基酸轉運蛋白(excitatory amino acid transporter, EAAT)**再攝取至神經末梢及鄰近星狀膠細胞(astrocyte);膠細胞將其轉為麩醯胺後再輸回神經元,使麩醯胺扮演「無活性傳遞物儲存池」的角色。
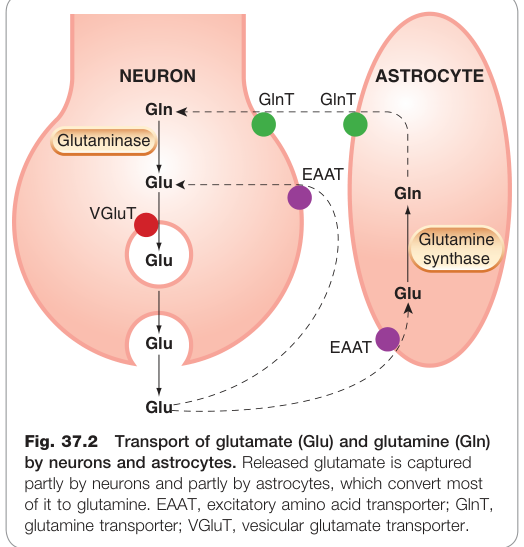
Figure 37.2:麩胺酸(Glu)與麩醯胺(Gln)在神經元與星狀膠細胞間的轉運循環
在缺血等病理狀態下,EAA 轉運蛋白可反向運作,將大量麩胺酸釋放至胞外,進而造成神經毒性(excitotoxicity)。
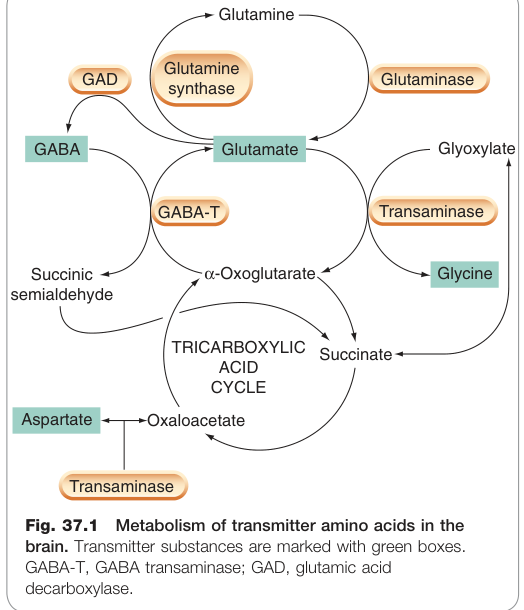
Figure 37.1:腦內傳遞物氨基酸的代謝途徑,包含 GAD(麩胺酸脫羧酶)與 GABA-T(GABA 轉胺酶)
麩胺酸受體亞型#
麩胺酸可活化**離子型(ionotropic)與代謝型(metabotropic)**兩大類受體。
離子型麩胺酸受體(Ionotropic Glutamate Receptors)#
依選擇性促效劑分為三種亞型,均為配體閘控陽離子通道:
| 受體 | 主要亞單元 | 通道特性 | 主要功能 |
|---|---|---|---|
| NMDA 受體(NMDA receptor) | GluN1、GluN2A–D、GluN3A–B | 慢動力學,高 Ca²⁺ 通透性 | 慢 EPSP、突觸可塑性、興奮毒性 |
| AMPA 受體(AMPA receptor) | GluA1–4 | 快動力學 | 快 EPSP,是腦功能運作的基礎 |
| 海藻酸受體(kainate receptor) | GluK1–5 | 快動力學,低 Ca²⁺ 通透性 | 快 EPSP、突觸前調節 |
NMDA 受體的特殊性質:
- 高 Ca²⁺ 通透性:活化後大量 Ca²⁺ 內流,觸發胞內訊號串聯。
- Mg²⁺ 電壓依賴性阻斷:靜息電位下 Mg²⁺ 封堵通道;持續去極化後阻斷解除。
- 需要甘胺酸為共促效劑:麩胺酸與甘胺酸均須結合才能開啟通道(Johnson 與 Ascher 的發現)。D-絲胺酸(D-serine)亦可活化甘胺酸結合位。
- 多胺調節:精胺(spermine)等內源性多胺可促進通道開啟;ifenprodil 可阻斷此作用。
- 通道阻斷劑:氯胺酮(ketamine)、苯環啶(phencyclidine)與 dizocilpine(MK801)均為 NMDA 通道開放狀態阻斷劑。

Figure 37.6:甘胺酸對 NMDA 受體的促進作用,顯示甘胺酸作為 NMDA 共促效劑的電生理證據
代謝型麩胺酸受體(Metabotropic Glutamate Receptors, mGlu)#
- 共八種(mGlu1–8),屬 C 類 G 蛋白偶合受體,以雙體(homodimer)形式運作,具有特殊的「捕蠅草(venus fly trap)」胞外結合域。
- 分為三組:
- 第 1 組(mGlu1、mGlu5):透過 Gq 蛋白升高胞內 Ca²⁺,主要位於突觸後,具興奮性調節作用。
- 第 2 組(mGlu2、mGlu3)與第 3 組(mGlu4、mGlu6–8):透過 Gi/Go 蛋白抑制突觸傳遞與神經元興奮性,多位於突觸前(可作為自體受體或異體受體)。
突觸可塑性與長期增益#
LTP 的機制(以海馬迴為例):
- 低頻活動時:僅 AMPA 受體活化,NMDA 通道被 Mg²⁺ 阻斷。
- 高頻刺激後:持續去極化移除 Mg²⁺ 阻斷,NMDA 受體開啟,Ca²⁺ 大量內流。
- 胞內 Ca²⁺ 升高活化 CaMKII(Ca²⁺/鈣調蛋白依賴性蛋白激酶)、PKC 等激酶,磷酸化 AMPA 受體、促其插入突觸膜,並透過 NO(一氧化氮)與花生四烯酸等**逆向傳訊物(retrograde messenger)**增強突觸前麩胺酸釋放。
- 維持期:基因表達改變,突觸結構重塑,突觸接觸數目永久增加。
- 長期抑壓(long-term depression, LTD):低頻刺激引發的適度 Ca²⁺ 內流活化磷酸酶,減少 AMPA 受體磷酸化,使突觸傳遞減弱。
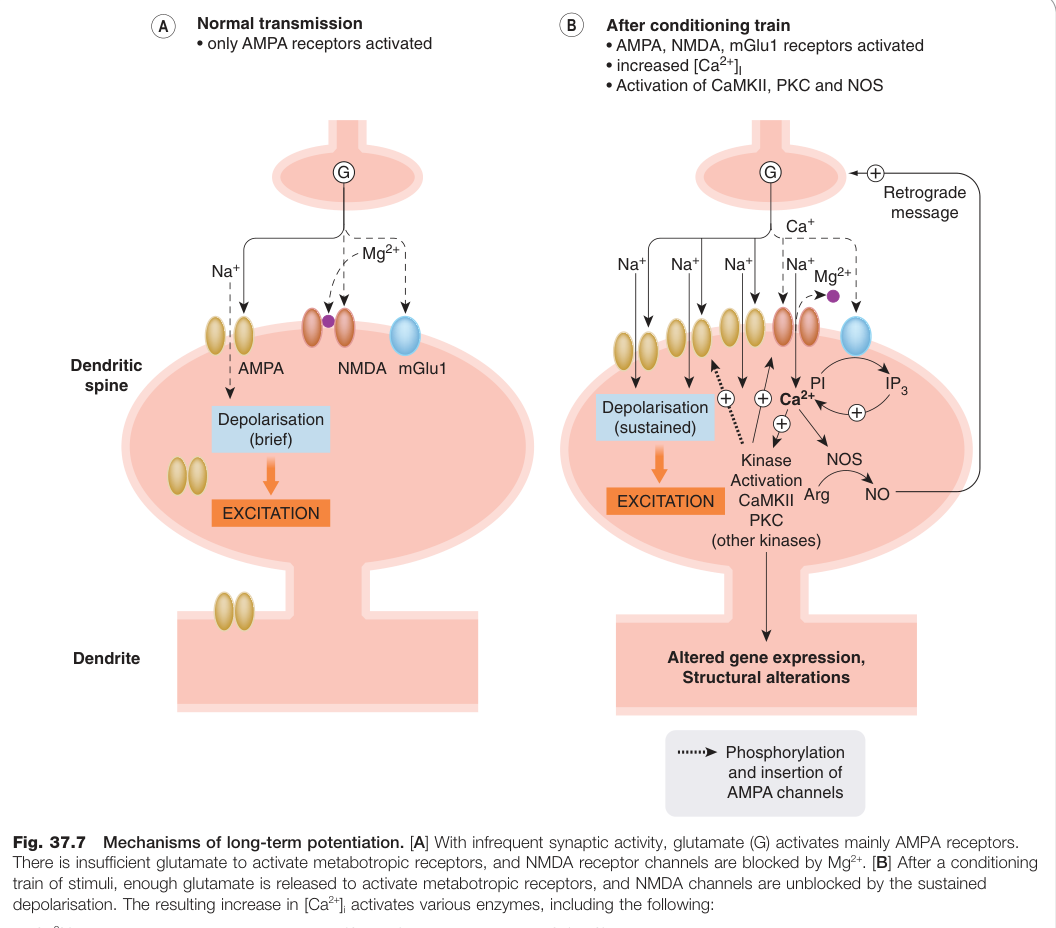
Figure 37.7:長期增益(LTP)的分子機制,顯示低頻活動時 AMPA 受體為主與高頻刺激後 NMDA 受體啟動的差異
作用於麩胺酸受體的藥物#
拮抗劑與負向調節劑#
- NMDA 受體競爭性拮抗劑:AP-5、CPP(實驗工具,無法穿越血腦屏障)。
- 甘胺酸位點拮抗劑:7-氯犬尿胺酸(7-chlorokynurenic acid)等,副作用或較少。
- NMDA 通道阻斷劑:ketamine(麻醉/鎮痛)、memantine(阿茲海默症,第 39 章)已進入臨床;dizocilpine、phencyclidine 為實驗藥物。
- AMPA 受體拮抗劑(如 NBQX):廣泛抑制 CNS,治療空間狹窄。
- GluK1 海藻酸受體拮抗劑:對疼痛、偏頭痛、癲癇等有潛力。
NMDA 拮抗劑(尤其通道阻斷劑)普遍有誘發幻覺等精神症狀的缺點,限制了臨床應用。
促效劑與正向調節劑#
- Ampakines(如 cyclothiazide、piracetam、CX-516):正向調節 AMPA 受體,增強 LTP,被評估用於認知促進、思覺失調症與憂鬱症。
- 第 2、3 組 mGlu 促效劑(如 LY354740):減少麩胺酸釋放,具神經保護與抗癲癇潛力。
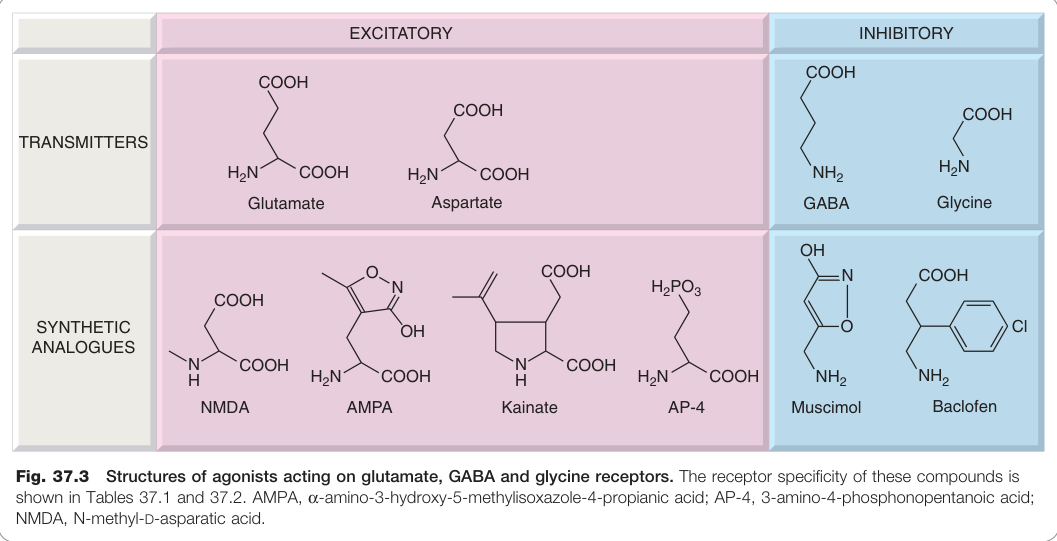
Figure 37.3:作用於麩胺酸、GABA 及甘胺酸受體的促效劑結構,包含 AMPA、NMDA、海藻酸等選擇性配體
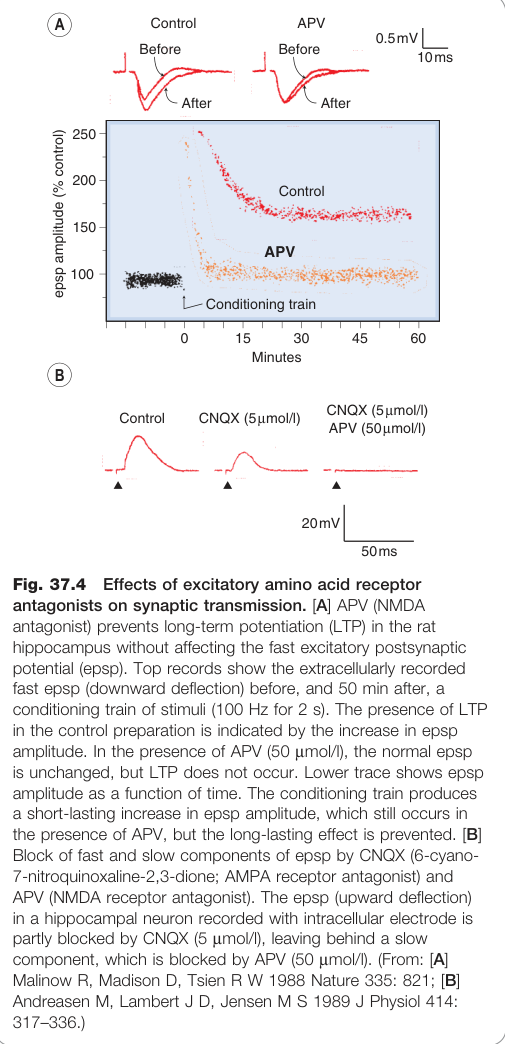
Figure 37.4:興奮性氨基酸受體拮抗劑對突觸傳遞的影響,包含 APV 阻斷 LTP 的電生理記錄
γ-胺基丁酸(GABA)#
合成、儲存與功能#
- GABA 是大腦主要的抑制性傳遞物,僅存在於腦組織(其他哺乳類組織含量極微)。
- 由麩胺酸經**麩胺酸脫羧酶(glutamic acid decarboxylase, GAD)**合成;GAD 只存在於 GABA 能神經元,可用於標記 GABA 路徑。
- 約 20% 的 CNS 神經元為 GABA 能;大多為短突觸間神經元(interneuron),也存在長距離 GABA 路徑(如紋狀體 → 黑質)。
- GABA 作用終止主要靠再攝取(轉運蛋白)及 GABA 轉胺酶(GABA transaminase)代謝降解。
拮抗劑 bicuculline 可誘發癲癇,顯示 GABA 對腦部持續的抑制作用不可或缺。
GABA 受體:結構與藥理#
GABA 作用於兩種受體:
GABA_A 受體#
- 屬 Cys-loop 家族,為五聚體配體閘控 Cl⁻ 通道(與甘胺酸受體、菸鹼受體、5-HT₃ 受體同族)。
- 已克隆 19 種亞單元(α1–6、β1–3、γ1–3、δ、ε、θ、π、ρ1–3),最常見組合為 α1β2γ2。
- GABA 結合於 α/β 亞單元界面;苯二氮平類(benzodiazepine)結合於 α/γ 界面。
- 活化後 Cl⁻ 內流,使細胞膜超極化,降低興奮性。
- 突觸外 GABA_A 受體(含 α4、α5、α6 及 δ 亞單元)對 GABA 親和力更高,去敏感化較慢,可被低濃度 GABA 持續活化,產生「緊張性抑制(tonic inhibition)」。
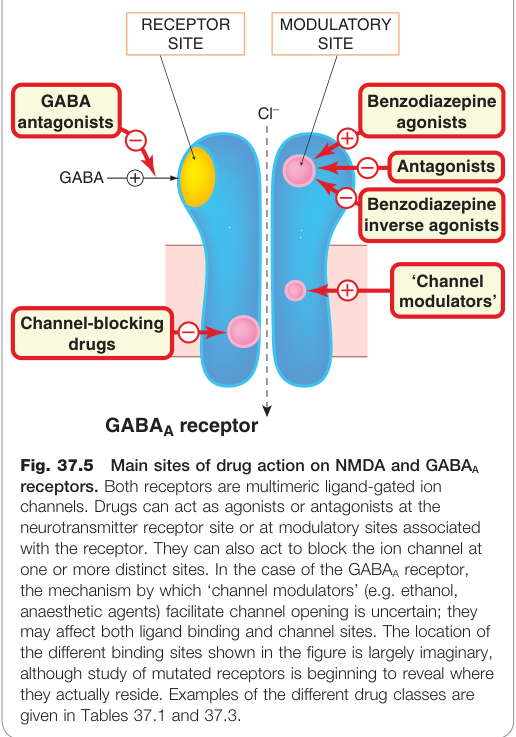
Figure 37.5:NMDA 與 GABA_A 受體的藥物作用位點,顯示促效劑、拮抗劑及多種調節劑(苯二氮平類、巴比妥類等)的結合位置
GABA_B 受體#
- C 類 G 蛋白偶合受體,以 **B1/B2 異二聚體(heterodimer)**形式發揮功能:
- B1 負責 GABA 結合;B2 與 G 蛋白偶合。
- 透過 Gi/Go 蛋白:抑制電壓閘控 Ca²⁺ 通道(減少突觸前傳遞物釋放)、開啟 K⁺ 通道(降低突觸後興奮性)、抑制腺苷酸環化酶。
- 分布於突觸前與突觸後。
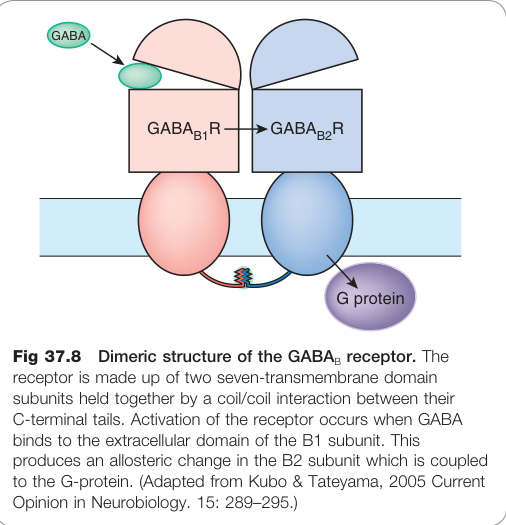
Figure 37.8:GABA_B 受體的雙聚體結構,由 B1(負責 GABA 結合)與 B2(負責 G 蛋白偶合)亞單元透過 C 端 coil/coil 交互作用組成
作用於 GABA 受體的藥物#
GABA_A 受體藥物:
- 促效劑:muscimol(來自致幻菇類);gaboxadol 為偏好 δ 亞單元的部分促效劑。
- 拮抗劑:bicuculline、gabazine(實驗工具,無臨床用途)。
- 正向調節劑(增強 GABA 效應):
- 苯二氮平類(diazepam 等):作用於輔助位點,依亞單元組成選擇性增強 GABA 結合;flumazenil 為其拮抗劑。
- 巴比妥類(barbiturates)、全身麻醉劑、神經類固醇(neurosteroid):作用位點不同,但均增強 GABA_A 活性。
- 通道阻斷劑:picrotoxin 阻斷 Cl⁻ 通道,為致痙攣劑(實驗工具)。
GABA_B 受體藥物:
- 巴氯芬(baclofen):選擇性 GABA_B 促效劑,臨床用於痙攣與運動障礙,亦評估用於藥物成癮(第 48 章)。
- 競爭性拮抗劑(如 2-hydroxy-saclofen、CGP 35348):動物實驗顯示有抗癲癇(失神發作)及認知促進效應,臨床應用尚待確認。
γ-羥基丁酸(GHB)#
- γ-羥基丁酸(γ-hydroxybutyrate, GHB)是 GABA 合成的天然副產物。
- 作為娛樂性藥物具有欣快與去抑制效應,在多數國家屬違法物質。
- 被認為是 GABA_B 受體的弱部分促效劑,亦結合特異性 GHB 受體,藥理機制尚未完全闡明。
甘胺酸(Glycine)#
- 甘胺酸是脊髓與腦幹最主要的抑制性傳遞物,在脊髓灰質含量特別高(約 5 µmol/g)。
- 在脊髓運動神經元,外加甘胺酸可引發與抑制性突觸電位相同的超極化效應。
- 馬錢子鹼(strychnine):甘胺酸受體競爭性拮抗劑,阻斷突觸抑制電位,引起痙攣。
- 破傷風毒素(tetanus toxin):阻斷甘胺酸從抑制性中間神經元釋放,導致強直性痙攣(lockjaw)。
甘胺酸的抑制性傳遞角色,與其作為 NMDA 受體共促效劑的角色完全不同,切勿混淆。
甘胺酸受體結構#
- 屬 Cys-loop 家族,為五聚體配體閘控 Cl⁻ 通道,結構與 GABA_A 受體相似。
- 亞單元較簡單(α1–4、β),成人腦中主要形式為 α1β 組合。
- 目前無專一性治療藥物;苯二氮平類及麻醉劑等增強 GABA_A 活性的藥物,對甘胺酸受體亦有類似增強作用。
甘胺酸轉運蛋白#
- GlyT1:主要位於星狀膠細胞,廣泛分布於 CNS。
- GlyT2:位於甘胺酸能神經元,主要在脊髓、腦幹與小腦。
- 抑制 GlyT1 可升高胞外甘胺酸濃度,透過增強 NMDA 受體活性,被評估用於思覺失調症的治療;GlyT 抑制劑亦具鎮痛潛力。
結語#
過去數十年,氨基酸受體的研究是神經科學最活躍的領域,累積了大量分子與藥理資訊。製藥界投入大量資源開發促效劑、拮抗劑與調節劑,進行了眾多臨床試驗,但迄今尚未出現重大突破。主要挑戰在於麩胺酸與 GABA 系統的廣泛性與多功能性——任何非選擇性藥物難免影響正常的腦功能。未來的希望在於開發針對特定受體亞型的高選擇性配體或變構調節劑。