定義與概述#
藥物動力學(pharmacokinetics) 是指對藥物在體內各區域濃度隨時間變化進行測量與正式解析的學科,常被概括為「身體對藥物做了什麼」。這與藥效學(pharmacodynamics)(「藥物對身體做了什麼」)互為對照。
藥物動力學的研究焦點通常是血漿濃度(plasma concentration,Cp),因為靜脈採血方便,且血漿濃度被假設與靶細胞周圍細胞外液中的藥物濃度有一定關聯,這是**目標濃度策略(target concentration strategy)**的基礎。
對特定藥物,臨床上會以血漿濃度個別化給藥,在達到治療效果的同時最小化副作用,此方法稱為治療藥物監測(therapeutic drug monitoring,TDM)。
藥物動力學的應用#
- 藥物開發:理解藥物在臨床前毒性試驗與動物藥理試驗中的行為
- 藥品監管:評估生物等效性(bioequivalence),決定學名藥上市
- 臨床給藥:制定並調整給藥方案,特別是腎臟或肝臟功能受損的患者
- 藥物交互作用:識別與評估可能的交互作用(見第 56 章)
清除率#
基本概念#
清除率(clearance,CL) 的概念源自 1929 年對尿素排泄率的表達方式——每分鐘被清除尿素的血量。藥物清除率類比定義為:每單位時間內,從體內移除所有藥物分子所需的血漿容積。
腎臟清除率(renal clearance,CLren) 的計算公式為:
$$CL_{ren} = \frac{C_u \cdot V_u}{C_p} \tag{10.1}$$
其中 $C_u$ 為尿液中藥物濃度,$V_u$ 為尿流速,$C_p$ 為血漿濃度。
總清除率(CLtot) 是描述藥物消除的根本藥物動力學參數:
$$\text{藥物消除速率} = C_p \times CL_{tot} \tag{10.2}$$
總清除率 = 腎臟清除率(CLren)+代謝清除率(CLmet)+其他途徑
清除率的測量方法#
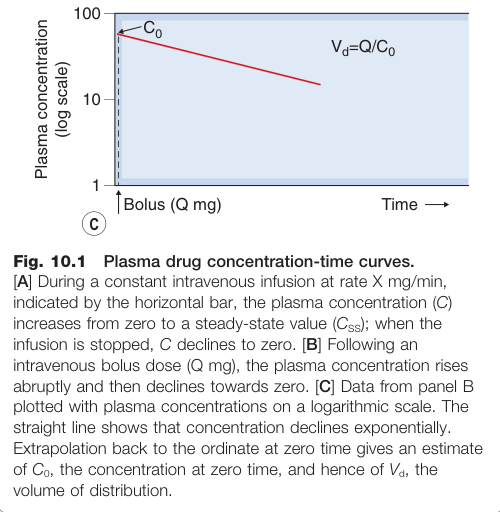
Figure 10.1:血漿藥物濃度—時間曲線,顯示靜脈滴注與單次靜脈注射給藥後的濃度變化及穩態(Css)的達成
方法一:穩態靜脈滴注
持續靜脈滴注(輸入速率 X mg/h)至穩態後,稳态濃度 $C_{SS}$ 滿足:
$$C_{SS} = \frac{X}{CL_{tot}} \tag{10.3,10.4}$$
方法二:單次靜脈注射(bolus)
$$CL_{tot} = \frac{Q}{AUC_{0-\infty}} \tag{10.5}$$
其中 $AUC_{0-\infty}$ 為濃度—時間曲線下面積(area under the curve)。
以 AUC 計算的清除率不依賴任何特定的房室模型,適用性較廣。
單房室模型#
模型架構#
單房室模型(single-compartment model) 將人體視為一個均勻混合的房室,體積為分布容積(volume of distribution,Vd)。藥物以靜脈注射方式進入,透過代謝或排泄離開。
給予單次靜脈注射劑量 Q 後,初始血漿濃度 $C_0$:
$$C_0 = \frac{Q}{V_d} \tag{10.6}$$

Figure 10.2:單房室藥物動力學模型示意圖,藥物在體內均勻分布,以 Vd 代表分布容積
一階動力學與半衰期#
大多數藥物遵循一階動力學(first-order kinetics),消除速率與濃度成正比,血漿濃度呈指數衰減:
$$C(t) = C(0) \cdot \exp!\left(-\frac{CL_{tot}}{V_d} t\right) \tag{10.7}$$
消除速率常數(elimination rate constant,kel) = $CL_{tot}/V_d$,單位為 (時間)$^{-1}$。
消除半衰期(elimination half-life,t₁/₂) 定義為血漿濃度降至 50% 所需時間:
$$t_{1/2} = \frac{\ln 2}{k_{el}} = \frac{0.693 \cdot V_d}{CL_{tot}}$$
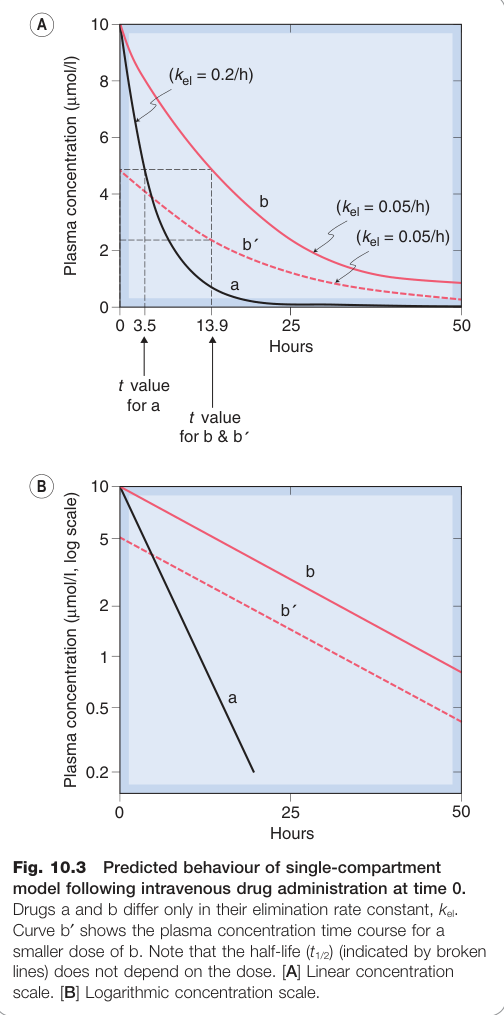
Figure 10.3:單房室模型靜脈注射後的預測血漿濃度曲線,比較不同消除速率常數 kel 的藥物行為
稳態濃度與重複給藥#
- 持續給藥後,血漿濃度以近似指數方式趨近穩態
- 實際上,給藥 3 ~ 5 個半衰期後即達到有效穩態
- 不論給藥頻率(單次大劑量或多次小劑量),平均穩態濃度相同
- 若臨床緊急需要快速達到治療濃度,可先給負荷劑量(loading dose)
負荷劑量的計算:
$$L = C_{target} \times V_d$$
負荷劑量的大小由 $V_d$ 決定,而非半衰期。
需要快速達到治療濃度的藥物(如胺碘酮 amiodarone、地高辛 digoxin、肝素 heparin)常使用負荷劑量。
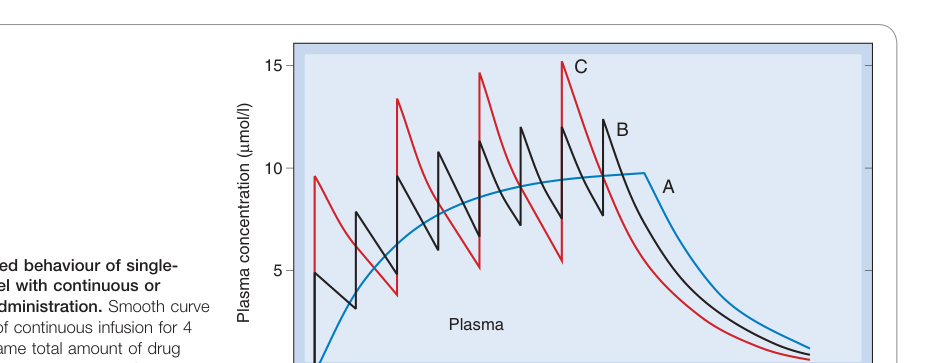
Figure 10.4:單房室模型在連續輸注(曲線 A)與分次等量給藥(曲線 B)下,血漿濃度隨時間變化的預測行為
吸收速率的影響#
若藥物從腸道或注射部位緩慢吸收,可用吸收速率常數 $k_{abs}$ 模擬:
- 吸收越慢,峰值濃度(Cmax)越低且出現越晚
- 吸收速率不影響 AUC(只要吸收完全)
- 吸收速率不影響穩態濃度,但影響濃度波動幅度
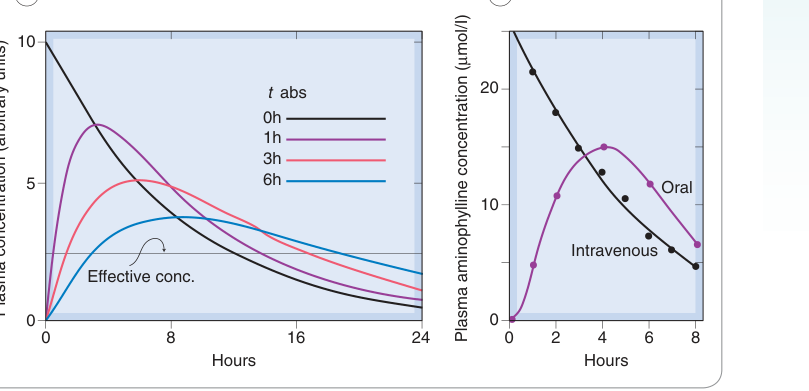
Figure 10.5:單房室模型中不同吸收速率對血漿藥物濃度的影響,消除半衰期固定為 6 小時
更複雜的動力學模型#
兩房室模型#
兩房室模型(two-compartment model) 將組織合併為一個外周房室(peripheral compartment),藥物只能經由中央房室(central compartment,通常代表血漿)進出外周房室。
兩房室模型預測的血漿濃度—時間曲線呈雙指數衰減:
- α 相(快相):藥物從血漿轉移至組織(再分布)
- β 相(慢相):藥物由體內消除

Figure 10.6:兩房室藥物動力學模型示意圖,中央房室(血漿)與外周房室(組織)之間的藥物轉移
地西泮(diazepam)是典型的兩房室藥物,在半對數圖上呈現明顯的雙相衰減:快相半衰期約 1.2 h,慢相半衰期約 30 h。
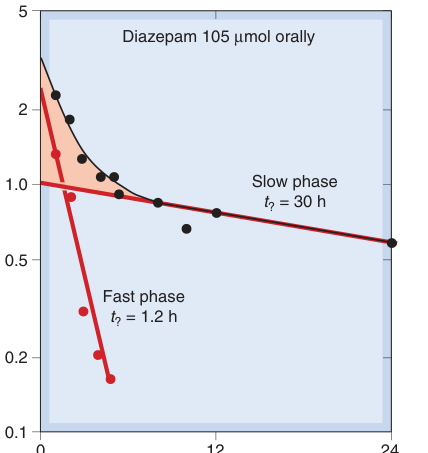
Figure 10.7:單次口服 diazepam 後人體血漿濃度的半對數圖,顯示約 8 小時後呈現線性的雙相消除動力學
若藥物代謝非常快速,α 相與 β 相不易分離,計算 Vd 與 kel 較複雜。極度親脂的藥物也不適合用單一「外周房室」概念描述。
飽和動力學(零階動力學)#
飽和動力學(saturation kinetics),又稱零階動力學(zero-order kinetics),出現在代謝酶被飽和的情形下。典型例子包括:
- 乙醇(ethanol):酒精去氫酶(alcohol dehydrogenase)受 NAD⁺ 供應限制,消除速率固定(約 4 mmol/L/h),與濃度無關
- 苯妥英(phenytoin):抗癲癇藥,代謝易飽和
- 水楊酸鹽(salicylate)
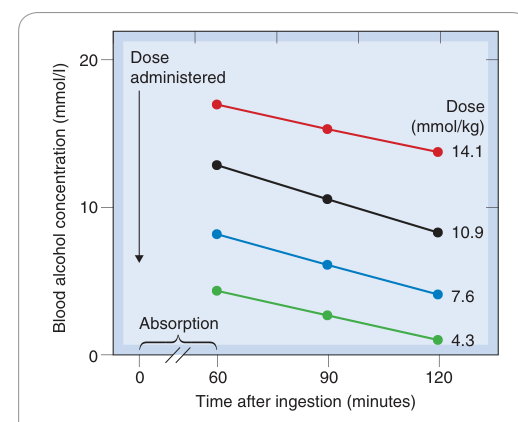
Figure 10.8:人體乙醇消除的飽和動力學,血中酒精濃度呈線性(非指數)下降,且下降速率不隨劑量改變

Figure 10.9:口服每 12 小時給藥下,非飽和線性動力學與飽和動力學(如苯妥英)的穩態濃度行為比較
飽和動力學的臨床後果:
- 劑量與穩態血漿濃度的關係陡峭且難以預測
- 若代謝速率超過最大代謝能力,藥物將在體內無限累積、永遠達不到穩態
- 酶誘導(enzyme induction)會造成血漿濃度不成比例的大幅變化
- 臨床用藥預測性遠低於線性動力學藥物
族群藥物動力學#
族群藥物動力學(population pharmacokinetics) 處理的情境是:無法在健康受試者上做標準研究(例如慢性病兒童患者),只能從臨床照護中零散採集稀疏數據。
常用方法:非線性混合效應模型(non-linear mixed effects modelling,NONMEM),可同時分析多個個體的稀疏數據,估計族群參數及個體間變異。
藥物動力學的局限性#
藥物動力學建立在兩個假設上,但均有例外:
血漿濃度準確反映靶位藥物濃度
- 對血液中作用的藥物(如溶栓藥物)成立
- 對需穿越血腦屏障(blood–brain barrier)的藥物則不然——抗憂鬱藥與抗精神病藥的血漿濃度監測並無臨床實用性
藥物反應僅取決於靶位藥物濃度
- 形成穩定共價鍵的藥物(如阿斯匹靈 aspirin、氯吡格雷 clopidogrel 的抗血小板作用)效果持續超過其在血漿中的存在時間
- 有時間延遲的藥物(如抗憂鬱藥)、或會誘導耐受性(opioids)或生理適應(皮質類固醇 corticosteroids)的藥物均不符合此假設
重點總結:
- $CL_{tot}$ 是藥物消除的根本參數;稳态濃度 $C_{SS} = \text{給藥速率}/CL_{tot}$
- 半衰期 $t_{1/2}$ 與 $V_d$ 成正比,與 $CL_{tot}$ 成反比
- 穩態在 3 ~ 5 個半衰期後達到
- 負荷劑量由 $V_d$ 決定:$L = C_{target} \times V_d$
- 兩房室模型呈雙指數動力學(α 相 + β 相)
- 飽和動力學藥物(苯妥英、乙醇)的穩態濃度隨劑量非線性升高,臨床難以掌控