概論#
藥物消除(drug elimination)是藥物從體內不可逆地消失的過程,涵蓋兩大機制:
- 代謝(metabolism):透過酵素將藥物化學轉化為其他物質
- 排泄(excretion):以原形藥物或代謝物的形式排出體外
主要排泄途徑:
- 腎臟(尿液)
- 肝膽系統(膽汁 → 糞便)
- 肺臟(僅限揮發性/氣態藥物,如全身麻醉藥)
脂溶性藥物無法由腎臟有效排除,因此需先在肝臟經細胞色素 P450(cytochrome P450,CYP)系統代謝為較具極性的產物,再由尿液排出。
藥物代謝#
動物演化出複雜的代謝系統以解除外來化學物質(xenobiotics)的毒性。藥物代謝分為兩個階段,通常依序發生,兩者皆會降低脂溶性、促進腎臟排除。
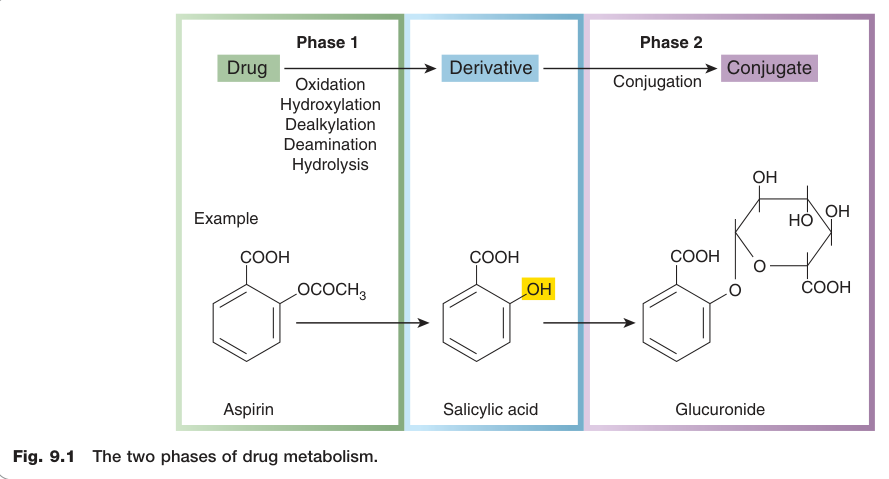
Figure 9.1:藥物代謝的兩個階段(Phase 1 與 Phase 2)概覽
第一相反應(Phase 1 Reactions)#
第一相反應屬分解代謝,包括氧化、還原與水解。
- 產物通常化學活性更高,有時比母藥更具毒性或致癌性
- 常見機制為「功能化(functionalisation)」:在分子中引入羥基等反應性基團,作為第二相共軛反應的攻擊位點
- 反應主要發生於肝臟的滑面內質網(smooth endoplasmic reticulum),相關酵素因分離純化時沉降於「微粒體分層(microsomal fraction)」而稱為微粒體酵素(microsomal enzymes)
脂溶性藥物可穿透細胞膜,進入細胞內接受代謝;極性藥物則較難進入,多以原形由尿液排出。
P450 單氧酶系統(P450 Monooxygenase System)#
P450 酵素的本質與分類
- 細胞色素 P450 為血基質蛋白(haem protein),由大家族的相關但相異酵素組成
- 命名規則:CYP + 數字(基因家族) + 字母(亞家族) + 數字(個別同功酶),如 CYP3A4
- 目前已描述 74 個 CYP 基因家族;CYP1、CYP2、CYP3 是人類肝臟中負責藥物代謝的三大家族
- 不同同功酶(isoenzyme)的受質特異性有重疊,但催化速率不同
主要同功酶與其受質範例:
| 同功酶 | 代表藥物 |
|---|---|
| CYP1A2 | 咖啡因、乙醯胺酚(→ NAPQI)、茶鹼 |
| CYP2C9 | 布洛芬、甲苯磺丁脲、華法林 |
| CYP2C19 | 奧美拉唑、苯妥英 |
| CYP2D6 | 可待因、去甲柔利、S-美托洛爾 |
| CYP2E1 | 酒精、乙醯胺酚 |
| CYP3A4/5/7 | 環孢素、硝苯地平、辛伐他汀 |
**反應機制:**單氧酶 P450 循環需要藥物(受質 DH)、P450 酵素、分子氧、NADPH 與黃素蛋白(NADPH–P450 還原酶)。淨效果為將分子氧中的一個氧原子加到藥物上形成羥基(DOH),另一個氧原子轉化為水。
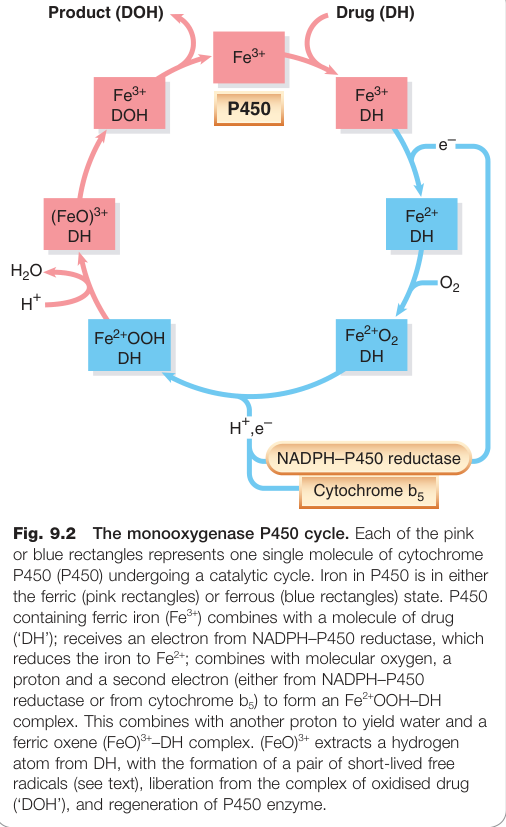
Figure 9.2:P450 單氧酶催化循環,顯示 Fe²⁺/Fe³⁺ 氧化還原狀態與藥物羥基化過程
非 P450 的氧化反應:
- 血漿膽鹼酯酶(hydrolysis of suxamethonium)
- 黃嘌呤氧化酶(xanthine oxidase):使 6-巰基嘌呤失活
- 單胺氧化酶(monoamine oxidase):使正腎上腺素、酪胺等胺類失活
- 醇脫氫酶(alcohol dehydrogenase):乙醇代謝(另兼 CYP2E1 途徑)
P450 的生物個體差異#
物種差異: 不同動物的 P450 表現與調控差異顯著,影響新藥毒性測試之物種選擇。
人類個體差異: 包括:
- 遺傳多型性(genetic polymorphisms):同一基因座的等位基因差異,跨世代持續存在於族群中(詳見第 11 章)
- 環境因素:飲食與環境中的酵素抑制劑或誘導劑(詳見第 56 章)
葡萄柚汁的成分可抑制 CYP 介導的藥物代謝,可能導致嚴重後果(如心律不整)。
抽菸、Brussels 芽甘藍則可誘導 P450 酵素,加速某些藥物代謝。
貫葉連翹(St John’s wort)可誘導多種 CYP450 同功酶及 P-醣蛋白(P-glycoprotein)。
P450 的抑制(Inhibition of P450)#
抑制劑依作用機制分類:
- 競爭性抑制:藥物競爭活性位點但本身不是受質(如奎尼丁競爭性抑制 CYP2D6)
- 非競爭性抑制:如酮康唑與 CYP3A4 血基質鐵(Fe³⁺)形成緊密複合物
- 機制性抑制(mechanism-based inhibition),又稱「自殺性抑制(suicide inhibition)」:需先被 P450 氧化,氧化產物再共價結合並破壞酵素本身(如口服避孕藥 gestodene 對 CYP3A4)
許多臨床重要的藥物交互作用源於 P450 酵素的抑制(詳見第 56 章)。
微粒體酵素的誘導(Induction of Microsomal Enzymes)#
部分藥物或化學物質(如利福平 rifampicin、乙醇、卡馬西平 carbamazepine、多環芳烴類致癌物)可大幅增加微粒體氧化酶與共軛酵素的活性。
- 機制:增加酵素合成和/或減少酵素分解
- 誘導機制類似類固醇及其他核受體配體的作用(見第 3 章)
- 多環芳烴(如 3-甲基膽蒽 3-MC)與 Ah 受體結合後,複合物轉運至細胞核,與 Ah 受體反應元件結合,促進 CYP1A1 轉錄
若藥物具有毒性代謝物(如乙醯胺酚),酵素誘導可能顯著增加藥物毒性與致癌性。
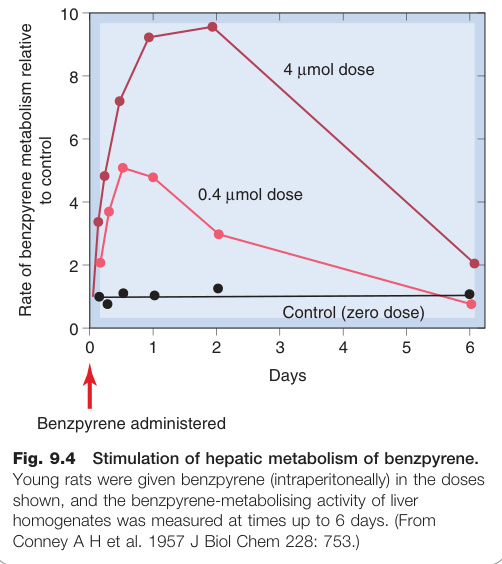
Figure 9.4:苯并芘(benzpyrene)誘導大鼠肝臟微粒體酵素活性的劑量效應
第二相反應(Phase 2 Reactions)#
第二相反應屬合成代謝(anabolic),以共軛(conjugation)為主要反應形式——在藥物分子中加上取代基。
- 通常產生無活性且極性較高的產物,易於腎臟排除
- 共軛基團包括:葡萄糖醛酸基(glucuronyl)、硫酸根(sulfate)、甲基(methyl)、乙醯基(acetyl)
- 麩胱甘肽(glutathione)可透過其硫氫基(sulfhydryl group)與藥物或其第一相代謝物共軛(如乙醯胺酚的解毒)
例外(仍具活性的第二相代謝物):
- 米諾地爾(minoxidil)的硫酸根代謝物仍具活性
- 嗎啡-6-葡萄糖醛酸(morphine-6-glucuronide)為嗎啡的活性代謝物,已被開發為止痛藥
葡萄糖醛酸化(glucuronidation): 高能磷酸化合物尿苷二磷酸葡萄糖醛酸(UDPGA)將葡萄糖醛酸基轉移至受質的 N、O 或 S 原子上,形成醯胺、酯或硫醇鍵。UDP-葡萄糖醛酸基轉移酶(UDP-glucuronyl transferase)受質特異性極廣,亦負責膽紅素及腎上腺皮質固醇等內源性物質的共軛。
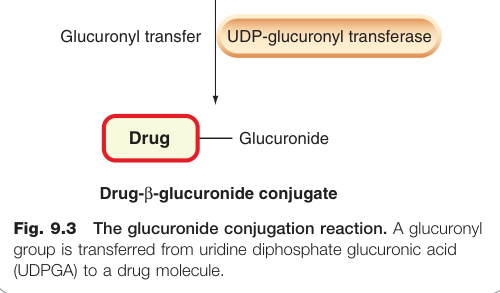
Figure 9.3:葡萄糖醛酸化共軛反應,UDPGA 將葡萄糖醛酸基轉移至藥物分子
立體選擇性(Stereoselectivity)#
許多臨床藥物為立體異構物混合物(如索他洛爾 sotalol、華法林 warfarin、環磷醯胺 cyclophosphamide),各異構物的:
- 藥理作用不同
- 代謝途徑可能完全不同
- 毒性可能主要由特定異構物引起
監管機構鼓勵新藥盡量開發為單一異構物(single isomer),以避免立體異構物相關的複雜性。
首過代謝(First-Pass / Presystemic Metabolism)#
部分藥物在經腸道吸收後,被腸壁或肝臟高效萃取,導致進入體循環的量遠少於吸收量,稱為首過代謝(first-pass metabolism)或系統前代謝(presystemic metabolism)。
問題在於:
- 口服劑量需遠高於注射劑量
- 個體間首過代謝的差異顯著
具顯著首過代謝的代表藥物: 阿斯匹靈、甘油三硝酸酯、左旋多巴、利多卡因、嗎啡、普萘洛爾、沙丁胺醇、維拉帕米等。
藥理活性代謝物(Pharmacologically Active Metabolites)#
部分藥物需先代謝才具藥理活性,稱為前藥(prodrugs):
| 前藥 | 活性代謝物 |
|---|---|
| 硫唑嘌呤(azathioprine) | 巰基嘌呤(mercaptopurine) |
| 皮質素(cortisone) | 氫皮質素(hydrocortisone) |
| 依那普利(enalapril) | 依那普利拉(enalaprilat) |
| 環磷醯胺(cyclophosphamide) | 磷醯胺氮芥(phosphoramide mustard) |
具毒性代謝物的藥物:
- 乙醯胺酚 → NAPQI(N-乙醯-對苯醌亞胺)→ 肝毒性
- 環磷醯胺 → 丙烯醛(acrolein)→ 膀胱毒性
- 甲醇、乙二醇 → 醇脫氫酶代謝物 → 嚴重全身毒性(治療:以乙醇競爭同一酵素)
膽汁排泄與腸肝循環(Biliary Excretion and Enterohepatic Circulation)#
肝細胞透過類似腎小管的轉運系統(OCT、OAT、P-醣蛋白)將藥物從血漿轉運至膽汁。
- 多種親水性藥物共軛物(尤其是葡萄糖醛酸結合物)濃縮於膽汁並輸送至腸道
- 葡萄糖醛酸在腸道中被水解,釋出活性藥物 → 重新吸收 → 循環反覆
- 此**腸肝循環(enterohepatic circulation)**可形成約佔體內總藥量 20% 的再循環「儲庫」,延長藥物作用時間
- 重要例子:嗎啡、乙炔雌二醇(ethinylestradiol)
維庫溴銨(vecuronium,非去極化肌肉鬆弛劑)主要以原形由膽汁排出。
利福平(rifampicin)則在腸道緩慢去乙醯化,去乙醯化型態不被腸道再吸收,最終多以此形式由糞便排出。
腎臟藥物排泄(Renal Excretion)#
腎臟對藥物的排除速率差異極大(從幾乎完全清除如青黴素,到極緩慢清除如地西泮 diazepam)。三個基本機制:
1. 腎絲球過濾(Glomerular Filtration)#
- 分子量 < 20,000 的藥物可自由通過腎絲球毛細管
- 與血漿白蛋白高度結合的藥物(如華法林約 98% 結合)過濾量大幅減少(僅 2% 可被過濾)
2. 腎小管主動分泌(Active Tubular Secretion)#
- 近端腎小管具有兩組相對非選擇性的載體系統:
- OAT(有機陰離子轉運體):轉運酸性藥物與尿酸
- OCT(有機陽離子轉運體):轉運有機鹼性藥物
- OAT 可逆電化學梯度主動運送,理論上可將血漿藥物濃度降至近零
- 由於至少 80% 的腎臟輸送藥物均接觸到載體,主動分泌是效率最高的腎排除機制
- 載體運輸不受蛋白結合率影響:游離藥物被轉運後,結合藥物解離補充,因此即使高度蛋白結合(如青黴素 80%),仍可被近乎完全清除
多種藥物競爭相同轉運系統而產生交互作用。丙磺舒(probenecid)即因抑制青黴素的腎小管分泌而被開發,用以延長青黴素的作用時間。
3. 跨腎小管被動擴散(Passive Diffusion Across the Renal Tubule)#
- 水分在腎小管中被重吸收(最終尿量僅為腎絲球濾液的約 1%)
- 若腎小管對藥物具有通透性,約 99% 的過濾藥物會沿濃度梯度被動重吸收
- 脂溶性藥物:容易重吸收 → 腎臟排除效率差
- 極性藥物(如地高辛 digoxin、胺基醣苷類抗生素):不易重吸收 → 在管腔中逐漸濃縮隨尿液排出
尿液 pH 與離子捕捉(Ion Trapping)#
- 許多藥物為弱酸或弱基,其游離度受 pH 影響,進而影響腎排泄速率
- 弱鹼性藥物(如安非他命)在酸性尿液中帶電,不易被重吸收 → 排泄加快
- 弱酸性藥物(如苯巴比妥、水楊酸鹽)在鹼性尿液中帶電 → 排泄加快
臨床上可透過尿液鹼化來加速水楊酸鹽(阿斯匹靈過量)的排除。
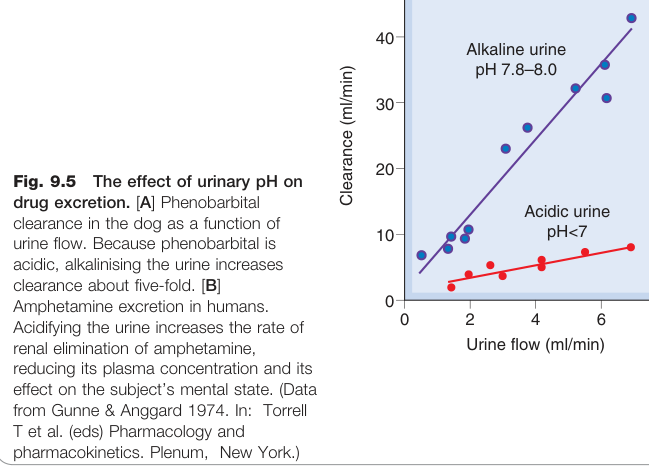
Figure 9.5:尿液 pH 對苯巴比妥(弱酸)與安非他命(弱鹼)腎臟排泄速率的影響
腎清除率(Renal Clearance)#
腎清除率 CLr 定義為單位時間內腎臟清除的血漿體積量:
$$CLr = \frac{C_u \times V_u}{C_p}$$
其中 $C_u$ = 尿液藥物濃度,$V_u$ = 尿流速,$C_p$ = 血漿藥物濃度。
CLr 範圍從 < 1 ml/min 到理論最大值約 700 ml/min(相當於腎血漿流速,以 p-胺基馬尿酸 PAH 清除率衡量)。
以腎臟排泄為主要消除途徑的藥物(如慶大黴素、甲胺蝶呤、地高辛),在老年人及腎功能受損患者中容易蓄積,需特別謹慎調整劑量。