概論#
藥物的吸收(absorption)與分布(distribution)取決於幾項關鍵物理過程:跨膜擴散、與血漿蛋白結合、以及分配進入脂肪與其他組織。本章也涵蓋各種給藥途徑,以及特殊藥物遞送系統的概念。
藥物在體內的處置(drug disposition)以縮寫 ADME 描述:
- Absorption(吸收):從給藥部位進入血液
- Distribution(分布):在體內各組織間分配
- Metabolism(代謝):由酵素轉化
- Excretion(排泄):從體內移除
藥物分子的物理移動#
藥物在體內以兩種方式移動:
- 整體流動(bulk flow):隨血流、淋巴或腦脊液移動
- 擴散(diffusion):分子逐一短距離移動
擴散速率主要取決於分子量(diffusion coefficient 與分子量平方根成反比),但多數藥物分子量落在 200–1000 Da 之間,此差異對藥動學影響有限。
跨細胞膜的路徑#
小分子穿越細胞膜有四條主要途徑:
- 直接通過脂質層(被動擴散)
- 通過水性孔道(aquaporins)——孔徑約 0.4 nm,對多數藥物太小
- 溶質載體(SLC transporter)或其他膜轉運蛋白
- 胞飲(pinocytosis)——主要用於大分子,如胰島素穿越血腦屏障
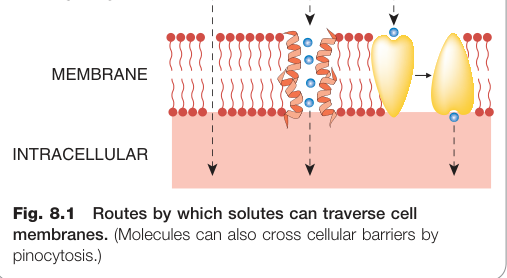
Figure 8.1:溶質穿越細胞膜的各種途徑(分子也可透過胞飲方式穿越)
脂溶性(lipid solubility)是決定被動跨膜速率最重要的因子,遠比分子量更關鍵。
穿越不同組織屏障#
不同組織的內皮結構差異很大:
- 一般毛細血管:細胞間有間隙,分子大小切斷點約 80,000–100,000 Da
- 中樞神經系統(CNS)與胎盤:緊密連接(tight junctions)+ 周細胞(pericytes),形成高度不通透的屏障
- 肝臟與脾臟:不連續內皮,允許分子自由通過
- 內分泌腺:有孔內皮(fenestrated endothelium),促進激素分泌
脂溶性與分配係數#
藥物穿越細胞膜的速率由 滲透係數 P(permeability coefficient)決定,P 取決於:
- 分配係數(partition coefficient):藥物在膜相與水相之間的分布比例
- 擴散係數(diffusion coefficient):藥物在脂質中的移動能力
由於不同藥物間擴散係數差異不大,分配係數(即脂溶性指標)就成為最關鍵的變數(見圖 8.2)。
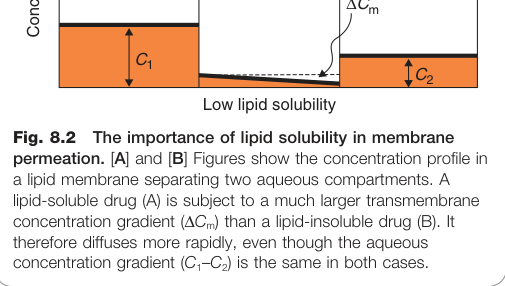
Figure 8.2:脂溶性對膜通透性的重要性,顯示脂溶性藥物(A)與非脂溶性藥物在脂質膜中的濃度分布
pH 與游離化(ionisation)#
許多藥物是弱酸或弱基,其游離比例依 Henderson–Hasselbalch 方程式隨 pH 變化:
- 弱鹼:
pKa = pH + log([BH⁺]/[B]) - 弱酸:
pKa = pH + log([AH]/[A⁻])
只有不帶電的游離形式才能穿越脂質膜。帶電的離子形式(BH⁺ 或 A⁻)脂溶性極低,幾乎無法穿越。
pH 分配與離子捕獲(ion trapping)#
當兩個體液區間存在 pH 差時,弱酸或弱基的藥物會因游離比例不同而在各區間濃度不均,稱為 pH 分配(pH partition):
- 弱酸傾向累積於高 pH 隔室(如鹼性尿液)
- 弱基傾向累積於低 pH 隔室(如胃液)
這一原則的臨床意涵:
- 阿斯匹靈(aspirin)中毒時,使用碳酸氫鈉(sodium bicarbonate)鹼化尿液可加速水楊酸鹽排泄;相反地,乙醯唑胺(acetazolamide)雖也鹼化尿液,卻會使水楊酸鹽更易進入 CNS,增加神經毒性
- 瘧疾藥物如氯喹(chloroquine)在寄生蟲酸性食物液泡中被捕獲,干擾血紅素分解
- 尿液酸化加快弱基排泄;鹼化則加快弱酸排泄
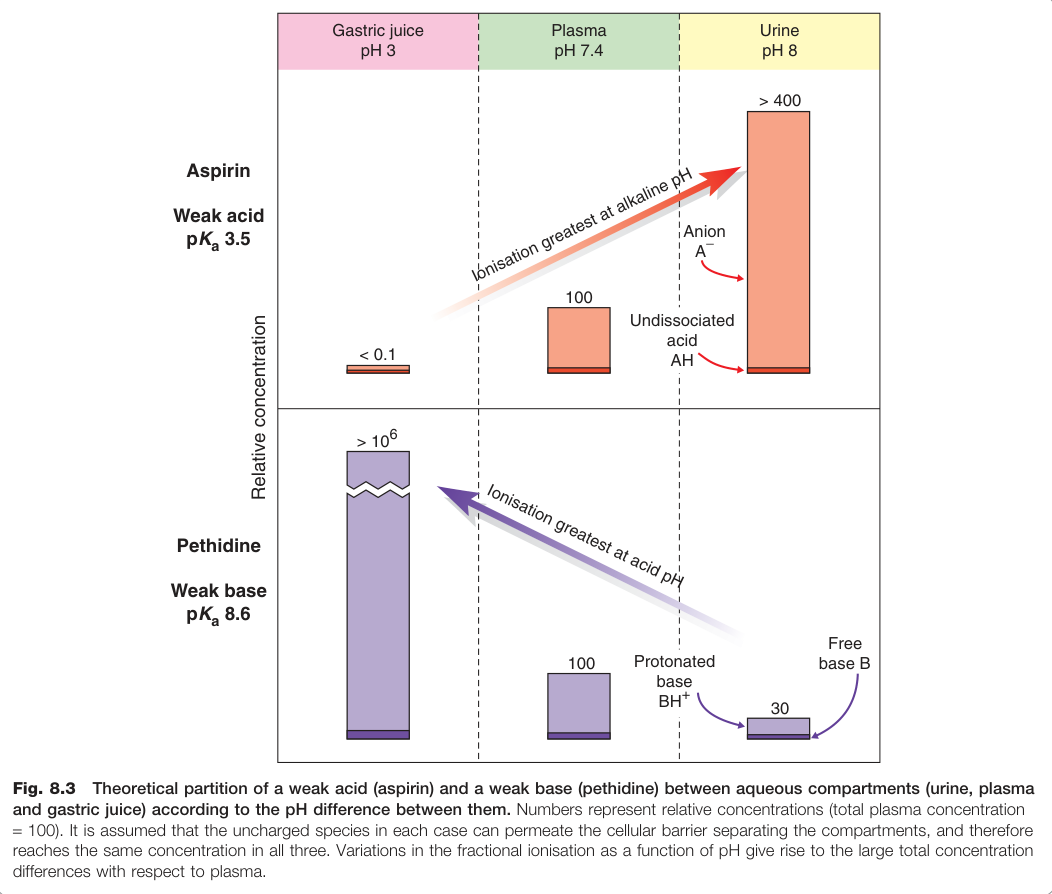
Figure 8.3:弱酸(阿斯匹靈)與弱基(配鉀啶)依 pH 差在尿液、血漿與胃液間的理論分配
pH 分配的理論梯度往往無法在現實中達到:一方面帶電形式並非完全不通透,另一方面胃液和腎小管液不斷流動,梯度無法達到平衡。此外,腸道吸收面積(絨毛)的差異才是決定吸收部位的主因。
載體介導的轉運#
細胞膜上的專一性轉運蛋白分兩大類:
- SLC 轉運體(solute carrier):促進分子順電化學梯度被動移動
- ABC 轉運體(ATP-binding cassette):利用 ATP 主動泵出分子
有機陽離子轉運體(OCT)與有機陰離子轉運體(OAT)#
- OCT:促進多巴胺、膽鹼等內源性物質及多種藥物(如 vecuronium、奎寧、普魯卡因胺)的擴散性轉運
- OCT2 位於腎臟近端小管,負責濃縮順鉑(cisplatin)→ 解釋其選擇性腎毒性;競爭抑制 OCT2 的西咪替丁(cimetidine)可提供保護
- OCT1 位於肝細胞,轉運二甲雙胍(metformin);OCT1 的單核苷酸多型性(SNP)會影響二甲雙胍療效
- OAT:負責尿酸、前列腺素、多種抗生素及抗病毒藥物的腎臟分泌
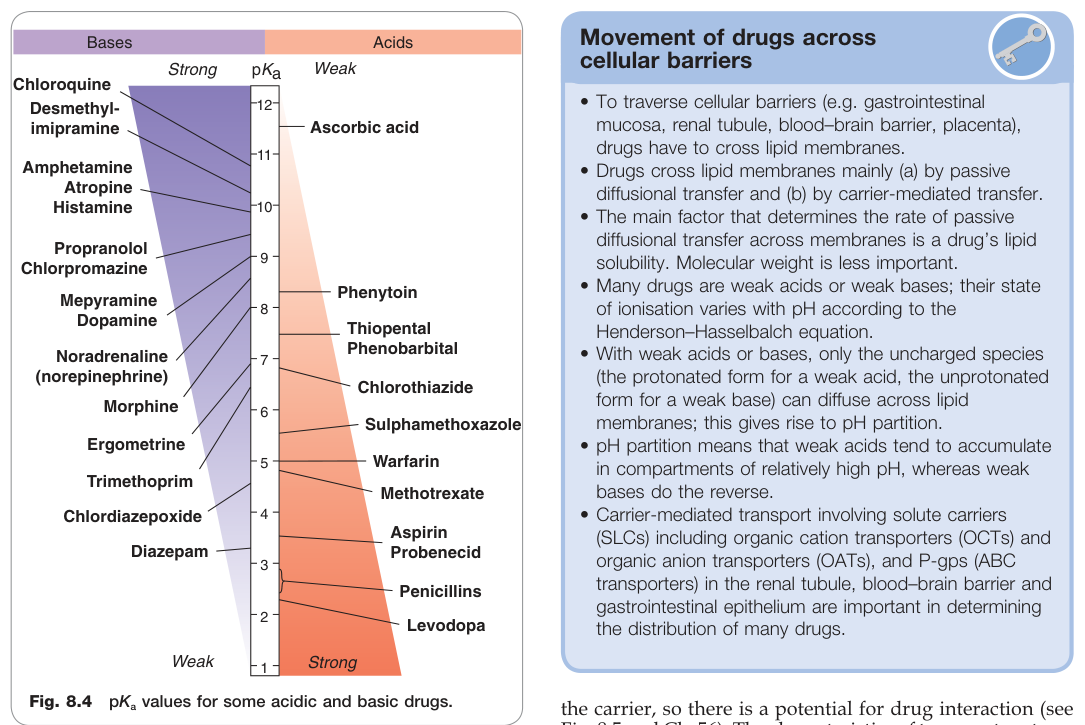
Figure 8.4:部分酸性與鹼性藥物的 pKa 值
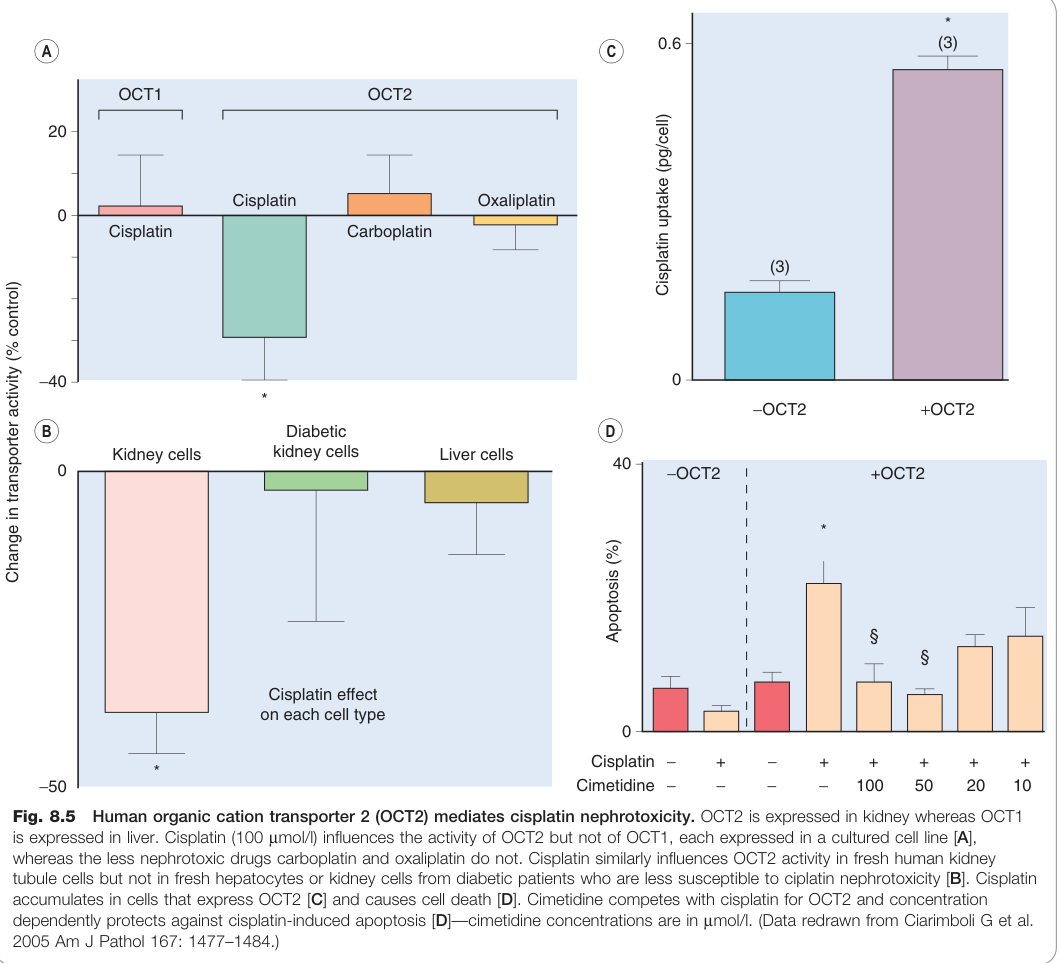
Figure 8.5:人類有機陽離子轉運體 OCT2 介導順鉑腎毒性,OCT2 表達於腎臟,OCT1 表達於肝臟
P-醣蛋白(P-glycoprotein, P-gp)#
P-gp 屬 ABC 轉運體,是癌細胞多藥抗藥性(multidrug resistance)的主因,分布於:
- 腎小管刷狀緣膜
- 膽管
- 腦微血管星形膠細胞突起(blood–brain barrier 的重要組成)
- 胃腸道
SLC 與 P-gp 常共同表達於同一組織。藥物可能先被 OAT 主動濃縮進細胞,再被 P-gp 泵出。SLC 與 P-gp 的遺傳多型性是個體間藥物反應差異的重要來源。
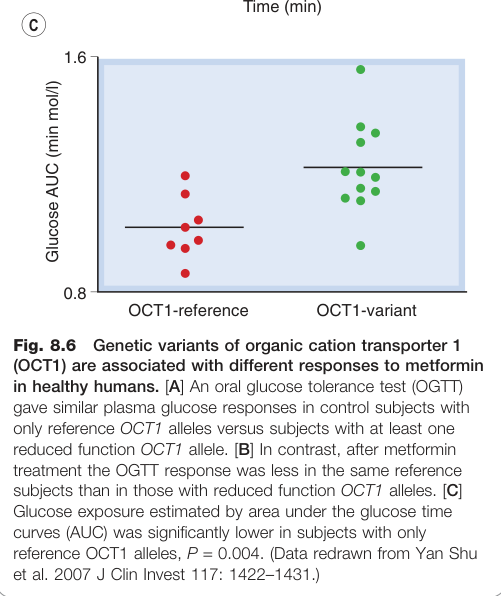
Figure 8.6:OCT1 遺傳變異與健康受試者對二甲雙胍(metformin)反應的差異
藥物與血漿蛋白的結合#
血漿中許多藥物以結合形式存在,游離藥物比例可低於 1%。只有游離的藥物才具有藥理活性。
- 最重要的血漿蛋白:白蛋白(albumin)
- 每個白蛋白分子約有 2 個結合位點,濃度約 0.6 mmol/L
- 主要結合酸性藥物(如 warfarin、NSAIDs、磺胺類)
- 也可結合部分鹼性藥物(如三環抗憂鬱藥、氯丙嗪)
- 其他蛋白:β-球蛋白和酸性醣蛋白(在發炎時增加)可結合某些鹼性藥物
結合量取決於三因素:
- 游離藥物濃度
- 與結合位點的親和力
- 蛋白質濃度
在一般治療濃度下,結合位點遠未飽和,結合分數基本固定。但少數藥物(如甲苯磺丁脲 tolbutamide)在接近飽和時,劑量加倍可導致游離濃度不成比例地大幅上升。
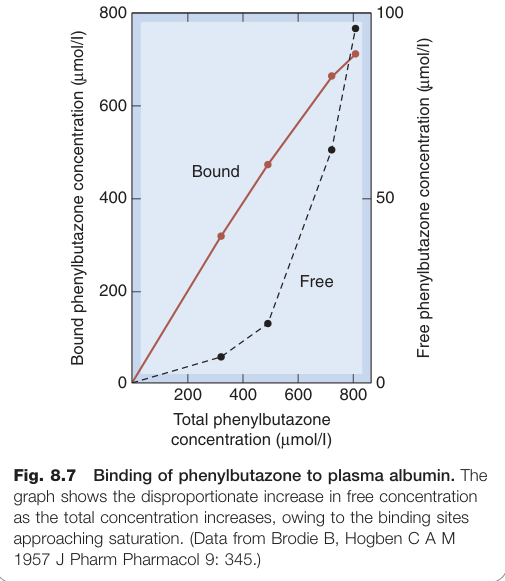
Figure 8.7:苯基丁氮酮(phenylbutazone)與血漿白蛋白結合,隨總濃度增加游離濃度不成比例上升(結合位點趨於飽和)
蛋白質結合的競爭#
當兩種藥物競爭同一白蛋白結合位點時,一種藥物可置換另一種,使其游離濃度上升。磺胺類藥物在治療濃度下佔據約 50% 的結合位點,可置換其他藥物或膽紅素(在早產兒中尤為危險)。
分配進入脂肪與其他組織#
- 脂肪:非極性大隔室,僅對高脂溶性藥物(如硫噴妥 thiopental,脂/水分配係數約 10)有顯著影響;硫噴妥因此迅速再分配進脂肪,限制其作為靜脈麻醉誘導劑的持續時間
- 脂肪血流量低(<2% 心輸出量),藥物蓄積緩慢;急性給藥時意義有限,長期服藥(如苯二氮平類 benzodiazepines)時則可顯著蓄積
- 其他特殊組織蓄積:
- 視網膜(黑色素):氯喹(chloroquine)→ 眼毒性
- 骨骼與牙齒(鈣):四環素(tetracyclines)→ 不可用於兒童
- 肝臟與肺:胺碘酮(amiodarone)長期使用 → 肝炎與肺纖維化
給藥途徑#
吸收(absorption) 定義為藥物從給藥部位進入血漿的過程,靜脈注射除外(F = 1 by definition)。
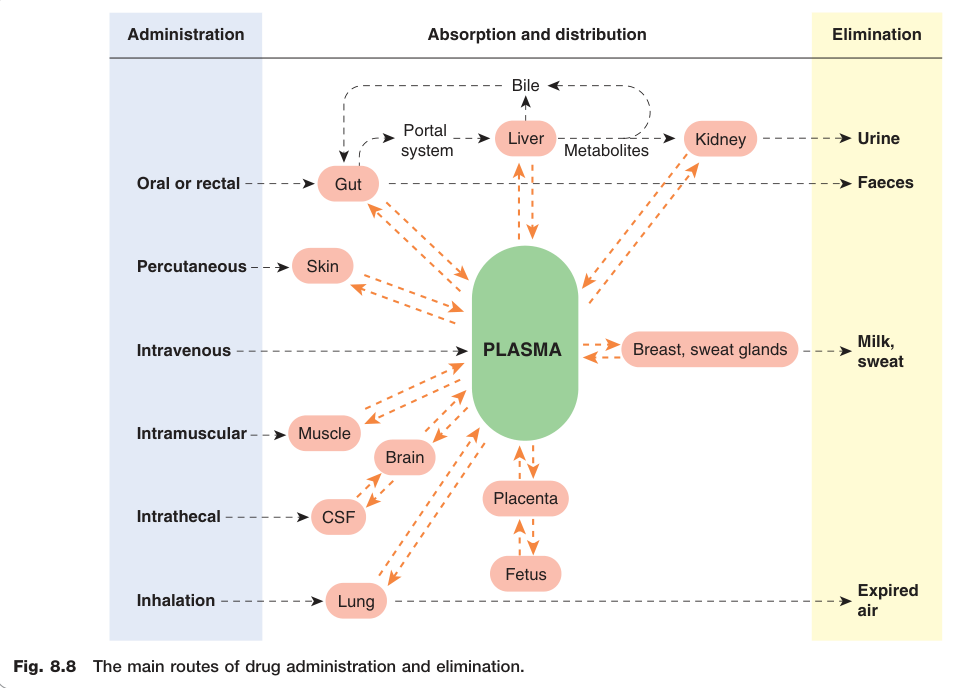
Figure 8.8:藥物給藥與排除的主要途徑
口服給藥#
多數藥物口服後在小腸(而非胃)吸收,因腸道絨毛的吸收面積遠大於胃。
腸道吸收機制#
- 弱酸(pKa < 3)或弱基(pKa > 10)的強電離化藥物吸收差
- 弱酸與弱基(pKa 在中間範圍)吸收良好
- 少數藥物依賴載體轉運(如 levodopa 透過苯丙胺酸轉運體;fluorouracil 透過嘧啶轉運體)
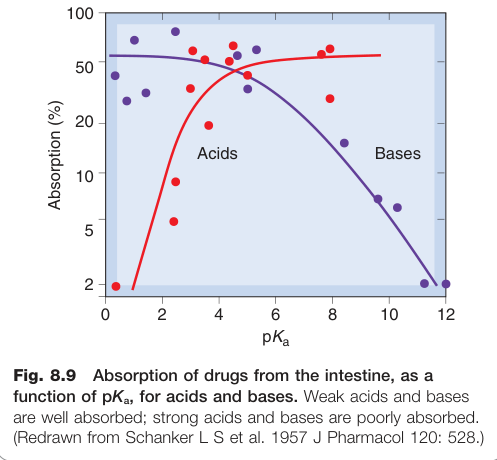
Figure 8.9:腸道藥物吸收與 pKa 的關係,弱酸與弱基吸收良好,強酸與強基吸收差
影響腸道吸收的因素#
- 胃腸蠕動:蠕動減慢(如偏頭痛、糖尿病神經病變)使吸收延遲;甲氧氯普胺(metoclopramide)加速胃排空而促進吸收
- 內臟血流:低血容量或心衰竭時血流減少,吸收降低
- 粒徑與劑型:粒徑差異可導致血漿濃度大幅不同(如不同廠牌地高辛錠的早期研究)
- 理化交互作用:四環素與鈣(如牛奶)結合而無法吸收;消膽胺(colestyramine)結合 warfarin 等藥物
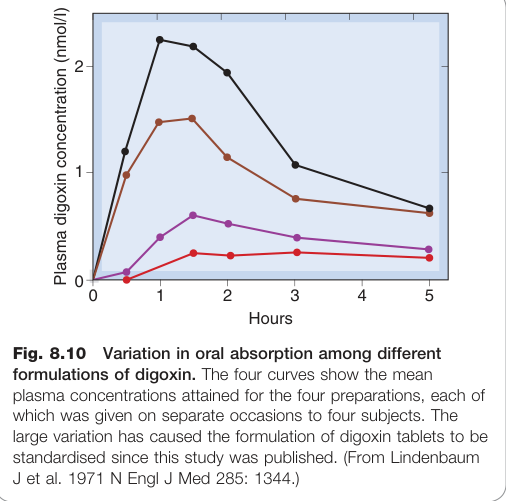
Figure 8.10:不同劑型地高辛(digoxin)口服吸收的差異,四種製劑給藥後的平均血漿濃度曲線
生體可用率(bioavailability)與生物等效性(bioequivalence)#
- 生體可用率(F):口服劑量中實際進入全身循環的比例,以 AUC_oral / AUC_iv 估算
- 影響 F 的因素:除吸收不完全外,首渡效應(first-pass metabolism)(腸壁及肝臟的前身代謝)也會降低 F
- 生物等效性:替代製劑的 AUC₍₀₋∞₎、Cmax 與 tmax 須在原廠產品的 80–125% 範圍內
生體可用率並非製劑的固定特性,腸道 pH、肝臟酶活性、腸胃蠕動等都會影響同一製劑在不同時間或不同個體的 F 值。
舌下給藥#
藥物從口腔黏膜直接吸收,繞過門脈系統,不受首渡代謝影響。適用於需要快速起效或在胃酸中不穩定的藥物,如硝酸甘油(glyceryl trinitrate)和丁丙諾啡(buprenorphine)。
直腸給藥#
可產生局部效應(如潰瘍性結腸炎用抗炎藥)或全身效應(如小兒癲癇持續狀態時給予 diazepam)。吸收常不穩定,適用於嘔吐或無法口服的患者。
皮膚給藥#
- 外用藥物主要產生局部效應,但部分仍可全身吸收
- 多數藥物難以穿透完整皮膚,但有機磷農藥(organophosphate insecticides)是例外
- 經皮貼片(transdermal patch):可達穩定給藥速率,避免首渡效應,適用脂溶性藥物(如雌激素、睪固酮、fentanyl)
鼻噴劑與眼藥水#
- 肽激素類似物(如抗利尿激素、降鈣素)可由鼻黏膜吸收
- 眼藥水(如 dorzolamide 治療青光眼)可達局部效果,但仍有部分全身吸收(如 timolol 引起支氣管痙攣)
吸入給藥#
- 揮發性麻醉藥透過肺部同時給藥與排除,可快速調控血漿濃度
- 呼吸道作用藥(如 salbutamol、beclometasone)可在肺部達高濃度同時減少全身副作用
- 化學修飾(如 ipratropium 為四級銨鹽)可減少全身吸收
注射給藥#
| 途徑 | 特點 |
|---|---|
| 靜脈注射(iv) | 最快、最確定;彈丸(bolus)注射達高峰濃度,持續滴注可避免高峰 |
| 皮下/肌肉注射 | 比口服快,但依注射部位與局部血流而異 |
| 鞘內注射(intrathecal) | 用於無法穿越血腦屏障的藥物(如 methotrexate、aminoglycosides 治療 CNS 感染、脊椎麻醉) |
| 玻璃體內注射(intravitreal) | 如 ranibizumab 用於濕性老年性黃斑病變 |
延遲吸收的方法#
- 加入腎上腺素(adrenaline/epinephrine)縮血管以延長局部麻醉藥效
- 胰島素與魚精蛋白(protamine)或鋅結合形成長效劑型
- 酯化類固醇或抗精神病藥(如 fluphenazine decanoate)提升油溶性,緩慢吸收
藥物在體內的分布#
體液隔室#
體內水分分布於四大隔室(佔體重百分比):
- 血漿(plasma water):約 5%
- 間質液(interstitial water):約 16%
- 細胞內液(intracellular water):約 35%
- 跨細胞液(transcellular water)(腦脊液、腹腔液等):約 2%
- 脂肪(fat):約 20%
各隔室中的藥物以游離與結合兩種形式共存,pH 決定弱酸/弱基的電荷形式。
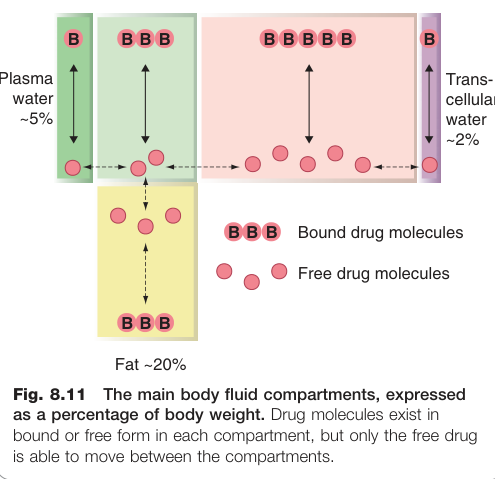
Figure 8.11:主要體液隔室佔體重的百分比,各隔室中藥物以游離與結合形式並存,只有游離藥物可在隔室間移動
表觀分布容積(Volume of Distribution, Vd)#
$$V_d = \frac{Q}{C_p}$$
其中 Q 為體內總藥量,Cp 為血漿濃度。Vd 是一個**表觀(apparent)**數值,不對應真實解剖隔室:
| Vd 範圍 (L/kg) | 意涵 | 代表藥物 |
|---|---|---|
| ~0.05 | 主要留在血漿 | heparin、insulin |
| ~0.1–0.2 | 強血漿蛋白結合 | warfarin |
| ~0.2 | 限於細胞外液 | vecuronium、gentamicin |
| ~0.55 | 分布全身體水 | ethanol、phenytoin |
| 2–5 | 廣泛組織分布 | morphine、propranolol |
| >10 | 大量組織結合 | nortriptyline、imipramine |
Vd 超過全身體積的藥物不能有效透過血液透析(haemodialysis)清除,因此藥物過量時血液透析通常無效。
血腦屏障(Blood–Brain Barrier, BBB)#
由 Paul Ehrlich 的染料實驗發現,BBB 由:
- 緊密連接的連續內皮細胞層
- 周細胞(pericytes)包覆
構成,使 CNS 不易受低脂溶性分子進入。
重要臨床意涵:
- 細菌性腦膜炎伴隨發炎可破壞 BBB,使靜脈注射青黴素得以進入 CNS
- 化學受體觸發區(chemoreceptor trigger zone)的屏障較鬆,允許 domperidone 作用於此而不進入基底核
- methylnaltrexone 因不穿越 BBB,可治療阿片類便秘而不影響中樞鎮痛效果
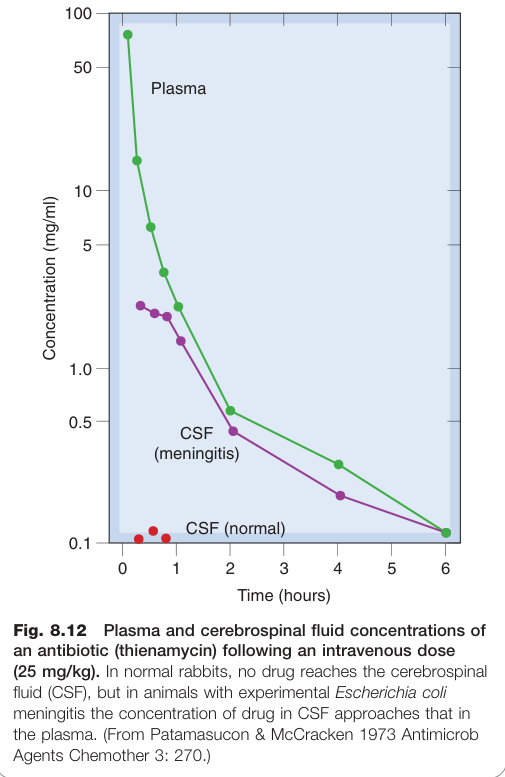
Figure 8.12:正常與腦膜炎兔靜脈注射抗生素(thienamycin)後血漿與腦脊液濃度比較,顯示 BBB 在發炎時通透性增加
特殊藥物遞送系統#
生物可降解奈米粒(Biologically Erodible Nanoparticles)#
高分子微球可黏附腸道黏膜,裝載高分子量藥物(如胰島素、質粒 DNA)提升口服吸收,應用仍在研究階段。
前驅藥(Prodrugs)#
前驅藥是無活性的前體,需在體內代謝後才轉化為有效形式:
- cyclophosphamide:在肝臟代謝後才具細胞毒性,可口服而不傷害胃腸道上皮
- levodopa:穿越 BBB 後在基底核轉化為 dopamine
- zidovudine:只在含適當逆轉錄酶的 HIV 感染細胞中磷酸化,具選擇性毒性
- valaciclovir / famciclovir:提升 aciclovir / penciclovir 的生體可用率
抗體–藥物結合物(Antibody–Drug Conjugates)#
將細胞毒性藥物連接至靶向腫瘤特異性抗原的抗體,提升癌症治療的選擇性。
脂質體(Liposomes)#
磷脂雙層微囊泡,可裝載水溶性或脂溶性藥物:
- 被網狀內皮系統(肝臟)攝取
- 在惡性腫瘤中累積
- 例:脂質體兩性黴素 B(liposomal amphotericin B)腎毒性低於傳統劑型
植入式緩釋裝置(Coated Implantable Devices)#
- 宮內裝置(IUD)對子宮內膜局部遞送荷爾蒙
- 冠狀動脈支架(stent)塗覆 sirolimus 等藥物,預防再狹窄(re-stenosis)