概覽#
本章從分子層面探討藥物如何辨識化學訊號並將其轉化為細胞反應。分子藥理學(molecular pharmacology)的進展正快速改變我們對藥物作用的認識,也開拓了大量新治療可能。
核心主題包含:
- 藥物作用的蛋白質靶點類型
- 四大受體超家族(receptor superfamily)的分子結構
- 受體與效應器的連結機制(訊號轉導)
藥物作用靶點#
哺乳類細胞上的蛋白質靶點可分為四大類:
- 受體(receptors)
- 離子通道(ion channels)
- 酵素(enzymes)
- 載體分子 / 轉運蛋白(carrier molecules / transporters)
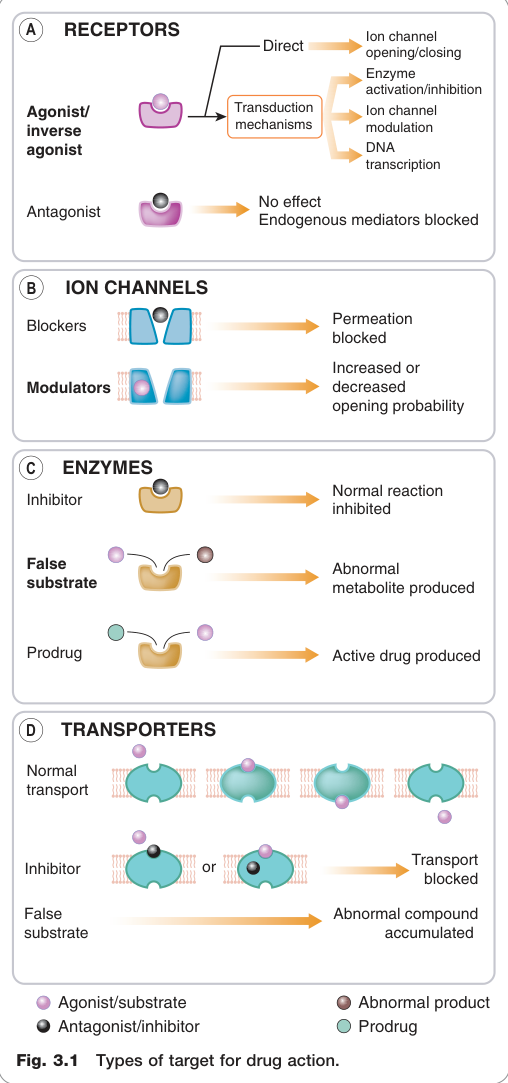
Figure 3.1:藥物作用的四大靶點類型
少數重要藥物作用於上述四類以外的蛋白質,例如秋水仙素(colchicine)作用於結構蛋白微管蛋白(tubulin),環孢素(ciclosporin)則作用於細胞質中的免疫親素(immunophilins)。
受體#
受體(receptor)是細胞化學通訊系統的感知元件。許多具有療效的藥物以已知內源性傳遞物質(endogenous mediator)的受體為靶點,扮演激動劑(agonist)或拮抗劑(antagonist)。在許多情況下,內源性傳遞物質比受體被發現更早;但也存在尚未確認配體的「孤兒受體(orphan receptor)」。
離子通道#
離子通道(ion channel)是細胞膜上的閘門,選擇性地允許特定離子通過。藥物可透過以下兩種方式影響離子通道:
- 直接結合:藥物分子嵌入通道(如局部麻醉藥物堵塞電位門控鈉通道),或結合通道上的變構位點(allosteric site)
- 間接作用:透過 G 蛋白(G-protein)及第二傳訊者(second messenger)等中間步驟調控通道功能
典型例子:
- **二氫吡啶類(dihydropyridines)**血管擴張藥:抑制 L 型鈣通道開放
- 苯二氮平類(benzodiazepines):結合 GABA_A 受體-氯離子通道複合體的調控位點,促進 GABA 開放通道
- 磺醯脲類(sulfonylureas):作用於胰臟 β 細胞的 ATP 門控鉀通道,促進胰島素分泌
酵素#
許多藥物以酵素(enzyme)為靶點:
- 競爭性抑制劑:如卡托普利(captopril)競爭性抑制血管緊張素轉換酶(angiotensin-converting enzyme)
- 不可逆結合:如阿司匹林(aspirin)不可逆地抑制環氧酶(cyclo-oxygenase)
- 假性受質(false substrate):如氟尿嘧啶(fluorouracil)取代尿嘧啶進入嘌呤生合成路徑,但無法轉化為胸苷酸,從而阻斷 DNA 合成
- 前體藥物(prodrug):需經酵素代謝才轉化為具活性形式
轉運蛋白#
轉運蛋白(transport protein)協助極性分子(無法自行穿透脂質膜的分子)穿越細胞膜。其識別位點也是藥物的作用標靶:
- ABC 轉運蛋白:如鈉泵(Na⁺-K⁺-ATPase)及多重抗藥性(MDR)轉運蛋白
- 離子耦合共轉運:神經傳遞物質轉運蛋白透過 Na⁺ 梯度驅動(同向轉運 symport 或反向轉運 antiport)
- 藥物範例:三環抗憂鬱藥(tricyclic antidepressants)、可卡因(cocaine)均透過阻斷去甲腎上腺素轉運蛋白(noradrenaline transporter)發揮作用
四大受體超家族#
根據分子結構與訊號轉導機制,受體可分為四種超家族:
| 類型 | 效應器 | 連結方式 | 時間尺度 | 代表例 |
|---|---|---|---|---|
| Type 1:配體門控離子通道 | 離子通道 | 直接 | 毫秒 | 菸鹼型乙醯膽鹼受體(nAChR)、GABA_A |
| Type 2:G 蛋白偶聯受體 | 酵素或通道 | G 蛋白 | 秒 | 毒蕈鹼型乙醯膽鹼受體(mAChR)、腎上腺素受體 |
| Type 3:激酶連結受體 | 蛋白激酶 | 直接 | 小時 | 胰島素受體、細胞激素受體 |
| Type 4:核受體 | 基因轉錄 | 經 DNA | 小時 | 類固醇荷爾蒙受體 |
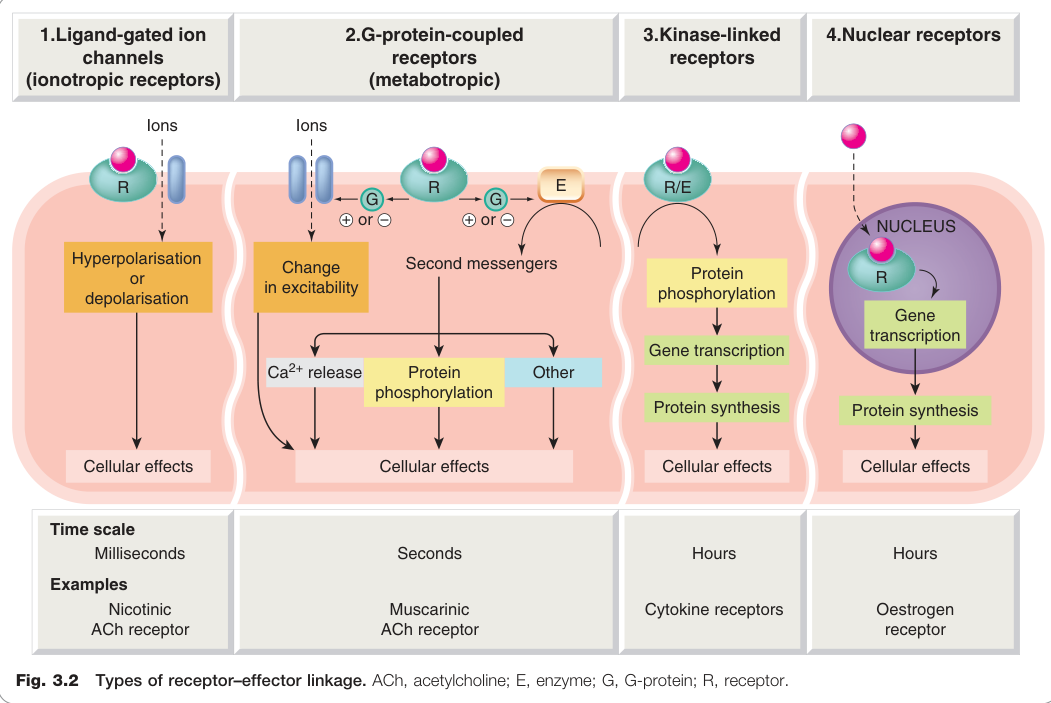
Figure 3.2:四大受體-效應器連結類型(ACh、G 蛋白、酵素 E、受體 R)
Type 1:配體門控離子通道#
分子結構#
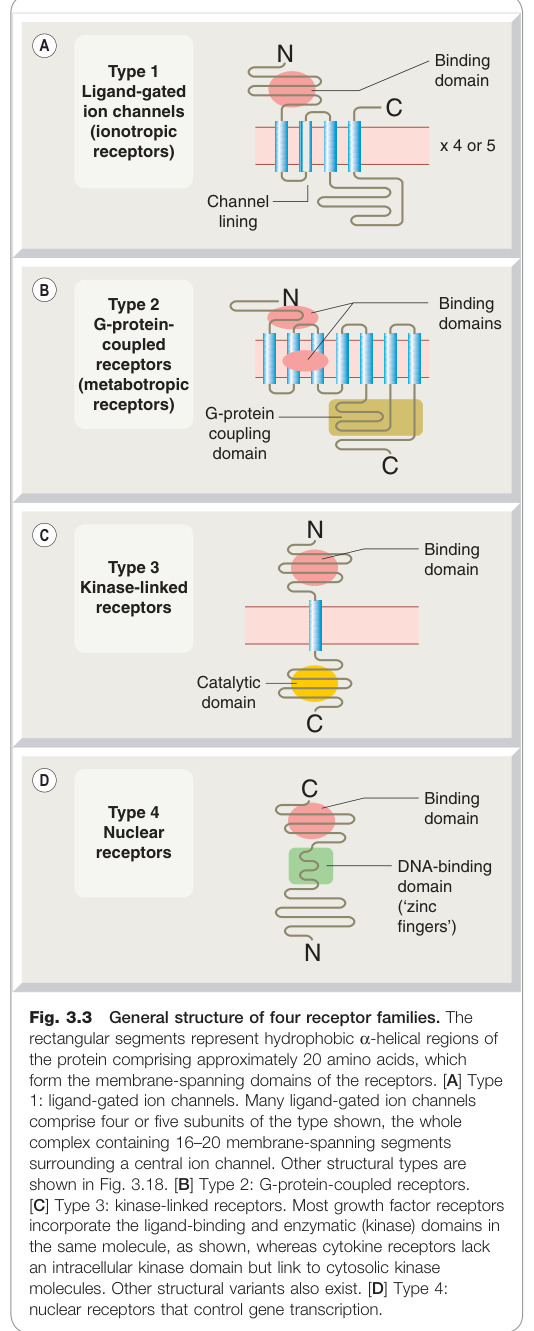
Figure 3.3:四大受體超家族的通用結構,矩形代表約 20 個胺基酸的疏水性跨膜 α-螺旋
- 由 4–5 個次單元(subunit)組成的寡聚體,圍繞中央親水孔道(pore)排列
- 菸鹼型乙醯膽鹼受體(nicotinic acetylcholine receptor, nAChR)是研究最深入的範例
- 五聚體(pentamer):α₂βγδ 各次單元,分子量 40–58 kDa
- 每個次單元含 4 條跨膜 α-螺旋(M1–M4);M2 螺旋構成孔道內壁
- 兩個乙醯膽鹼結合位點位於兩個 α 次單元的介面上,兩個位點均需結合才能激活受體
- 孔道內壁帶負電,對陽離子具選擇性通透
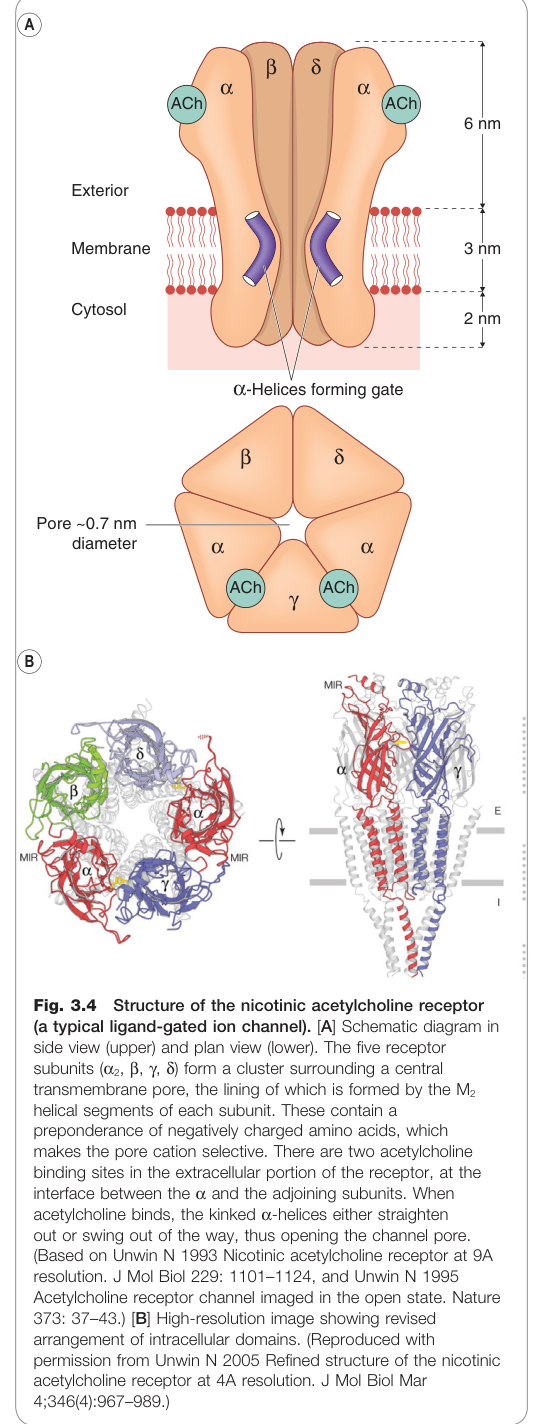
Figure 3.4:菸鹼型乙醯膽鹼受體(配體門控離子通道典型範例)的結構,包含側視與俯視示意圖
其他屬於此家族的受體(稱為 cys-loop 受體)包括 GABA_A、5-HT₃ 和甘氨酸(glycine)受體。麩胺酸(glutamate)受體(NMDA、AMPA、kainate 型)及辣椒素受體(TRPV1)則屬於不同的 P-loop 結構。
閘控機制#
配體門控離子通道控制神經系統中最快速的突觸事件,延遲僅數毫秒,不需要任何中間生化步驟,是受體與離子通道直接偶聯的典型。
- 興奮性神經傳遞物質(如乙醯膽鹼、麩胺酸)使 Na⁺/K⁺ 通透性上升,產生去極化
- Katz 與 Miledi(1972)的雜訊分析(noise analysis)技術,以及 Neher 與 Sakmann 的**膜片箝制(patch clamp)**技術,首次直接量測單一通道電流
- 單一通道電導:約 20 pS(相當於每秒約 10⁷ 個離子通過)
- 平均開放時間(mean open time):1–2 ms
- 不同激動劑產生相同的通道電導,但平均通道開放時間不同,反映各藥物的效能(efficacy)差異
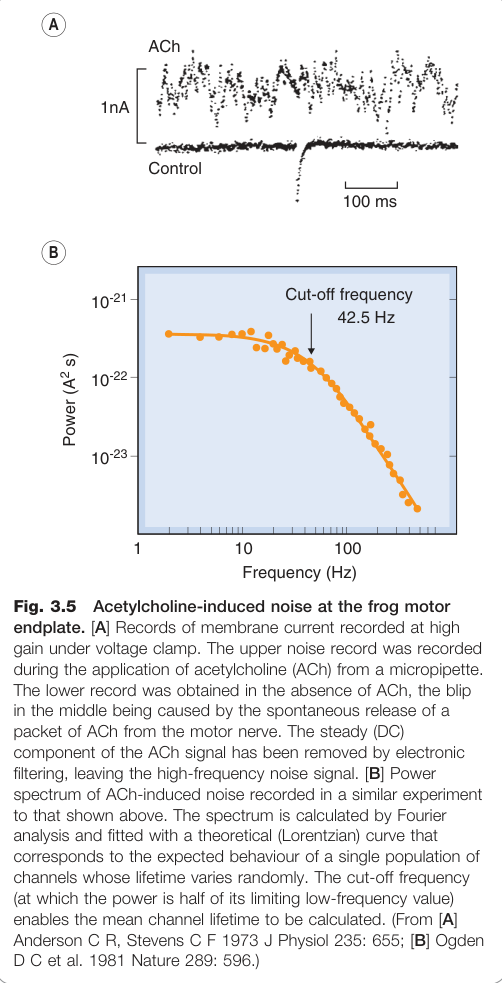
Figure 3.5:蛙運動終板的乙醯膽鹼誘發膜電流雜訊,用於分析單一通道電導
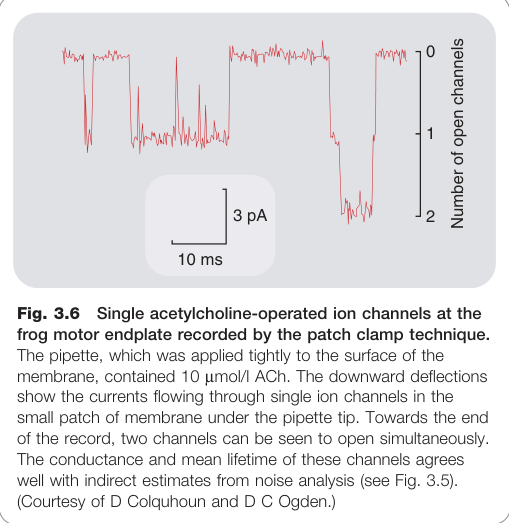
Figure 3.6:以膜片箝制技術記錄蛙運動終板單一乙醯膽鹼操控離子通道電流
配體門控離子通道的主要特徵:
- 又稱「離子促進型受體(ionotropic receptors)」
- 主要參與快速突觸傳遞
- 配體結合與通道開放在毫秒時間尺度完成
- 代表例:nAChR、GABA_A、5-HT₃
Type 2:G 蛋白偶聯受體#
分子結構#
- 人類基因組編碼約 400 個 GPCR(不含嗅覺受體),是治療藥物最常見的單一靶點類別
- 特徵結構:七個跨膜 α-螺旋(7-TM / heptahelical)
- 第三個細胞內迴圈(third cytoplasmic loop)負責與 G 蛋白偶聯
- 依家族分為三類:
- Family A(rhodopsin 家族):最大,包括大多數胺類神經傳遞物質受體、神經肽及大麻素受體;配體結合於跨膜螺旋間的裂縫中
- Family B(secretin/glucagon 家族):肽類荷爾蒙受體(降鈣素、升糖素等);配體結合於較長的胞外 N 端
- Family C(代謝型麩胺酸受體家族):成員最少,包括代謝型麩胺酸受體、GABA_B 受體及 Ca²⁺ 感知受體;具有很長的胞外配體結合區
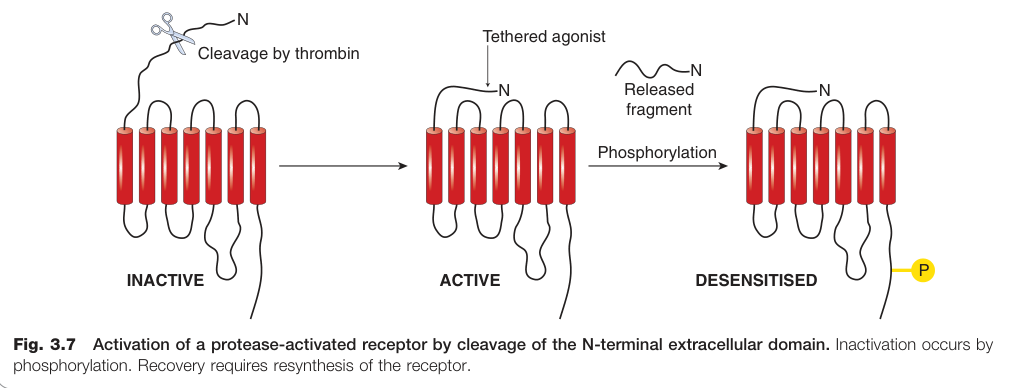
Figure 3.7:蛋白酶活化受體(PAR)的活化機制:N 端細胞外域被切割後暴露新配體序列,失活則由磷酸化驅動,恢復需重新合成受體
G 蛋白的功能#
G 蛋白(G-protein)由三個次單元組成:α、β、γ,其中:
- α 次單元具有 GTPase 活性(催化 GTP → GDP 水解)
- β 與 γ 次單元形成穩定的 βγ 複合體
- 靜止狀態:αβγ 三聚體,α 次單元結合 GDP
激活循環:
- 激動劑與 GPCR 結合,使受體構象改變
- αβγ 三聚體與活化受體結合,α 次單元釋放 GDP 並結合 GTP
- α-GTP 與 βγ 分離,兩者均可與下游效應蛋白結合
- α 次單元的 GTPase 活性水解 GTP → GDP,終止訊號
- α-GDP 重新與 βγ 結合,完成一個循環
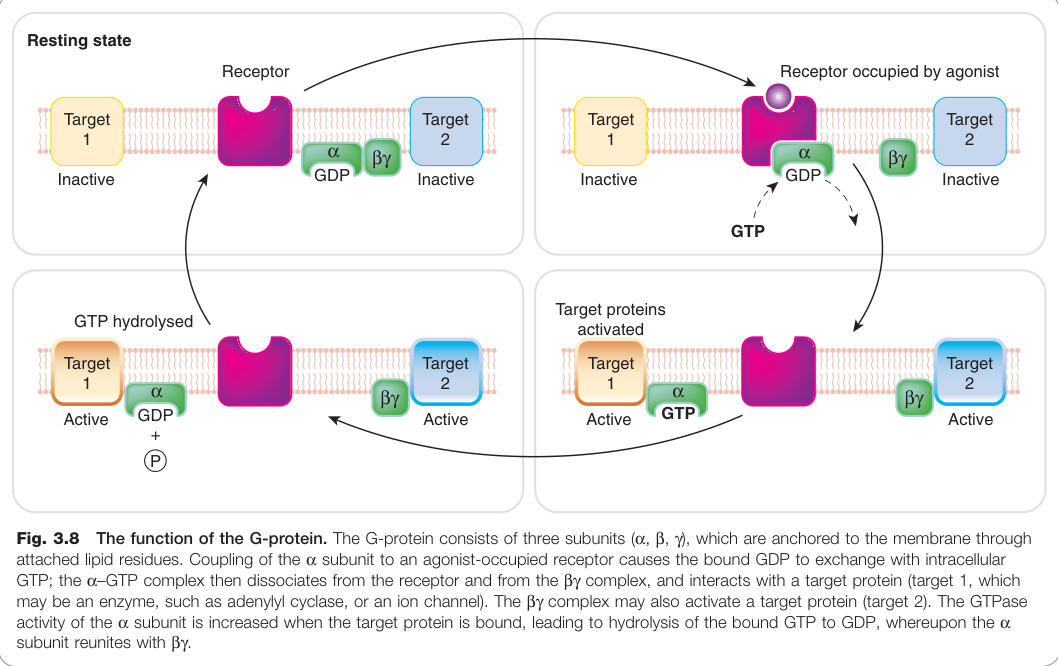
Figure 3.8:G 蛋白功能示意圖:α、β、γ 次單元透過脂質殘基錨定於膜上,激動劑活化受體後引發 GDP/GTP 交換與 α-GTP 分離
此機制可產生訊號放大效應:一個激動劑-受體複合體可依序活化多個 G 蛋白分子;每個 G 蛋白分子與效應酵素結合的時間足以產生大量產物。
主要 G 蛋白亞型:
| 亞型 | 偶聯受體 | 主要效應器 | 特殊性質 |
|---|---|---|---|
| Gα_s | 兒茶酚胺、組胺受體等 | 刺激腺苷酸環化酶,↑ cAMP | 可被霍亂毒素(cholera toxin)永久活化 |
| Gα_i | 阿片類、大麻素受體等 | 抑制腺苷酸環化酶,↓ cAMP | 可被百日咳毒素(pertussis toxin)阻斷 |
| Gα_q | 胺類、肽類受體 | 活化磷脂酶 C,↑ IP₃ 和 DAG | — |
| Gβγ | 所有 GPCR | 活化鉀通道、抑制鈣通道等 | 需較高的 GPCR 活化程度 |
G 蛋白控制的下游效應#
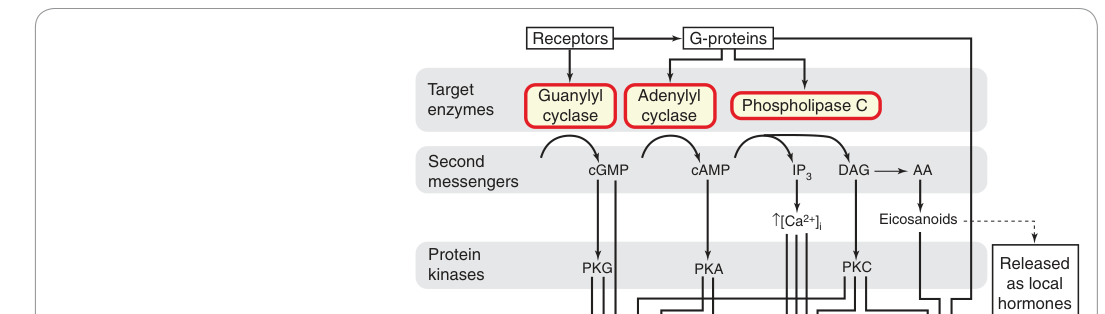
Figure 3.13:G 蛋白與第二傳訊者對細胞效應系統的調控總覽(AA,花生四烯酸;DAG,二醯甘油;IP₃,肌醇三磷酸)
腺苷酸環化酶 / cAMP 系統#
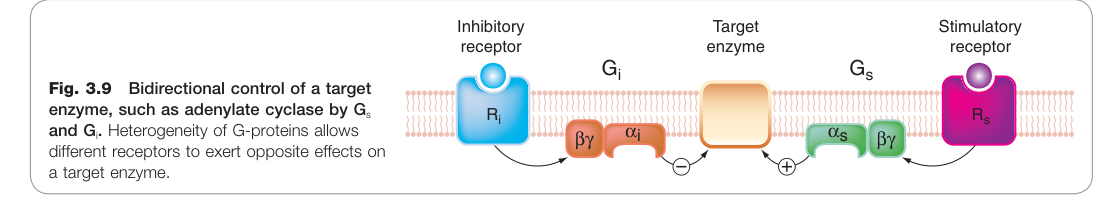
Figure 3.9:Gs 與 Gi 對目標酵素(如腺苷酸環化酶)的雙向調控,G 蛋白異質性使不同受體得以對同一效應酵素產生相反效果
- Gαs 活化腺苷酸環化酶(adenylyl cyclase)→ 合成 cAMP(環 3′,5′-腺苷一磷酸)
- cAMP 活化蛋白激酶 A(PKA)→ 磷酸化多種蛋白質(酵素、離子通道等)
- 磷酸二酯酶(phosphodiesterases, PDEs)水解 cAMP 終止訊號
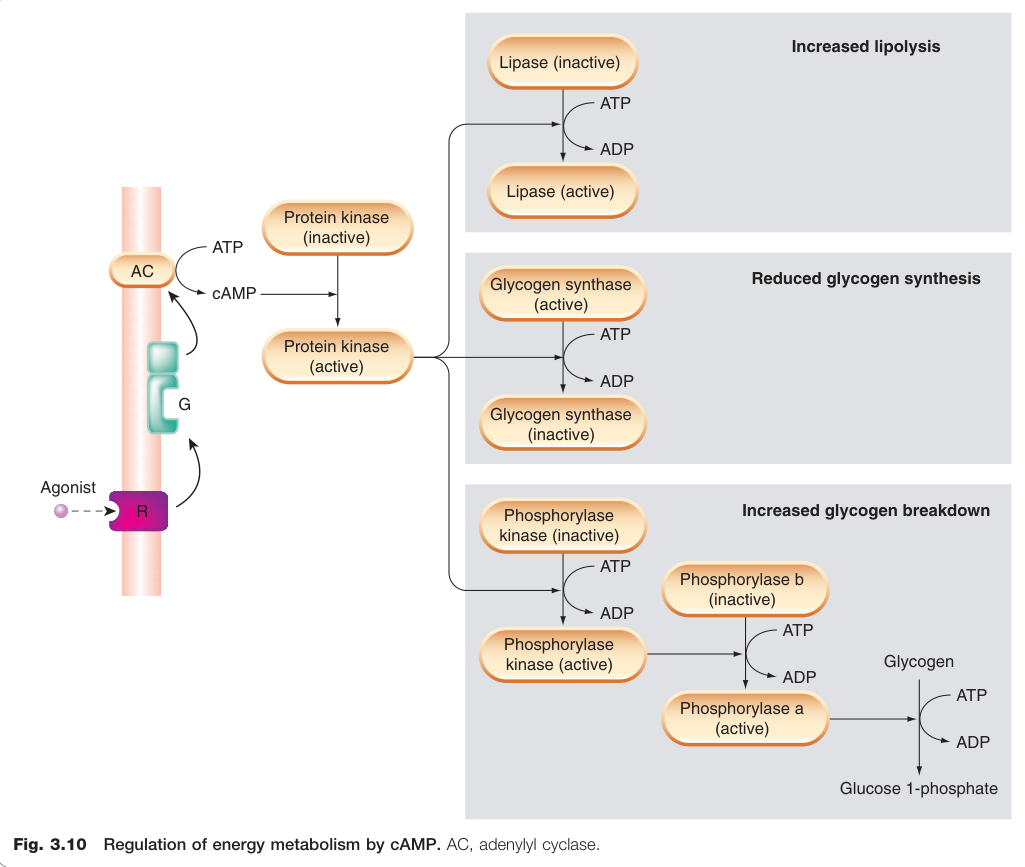
Figure 3.10:cAMP 調控能量代謝的路徑(AC,腺苷酸環化酶)
選擇性 PDE 抑制劑具重要臨床意義:
- 茶鹼(theophylline):廣泛抑制 PDE,用於氣喘
- 西地那非(sildenafil, Viagra):選擇性抑制 PDE5,增強一氧化氮的血管擴張效應(以 cGMP 介導)
- 米力農(milrinone):抑制 PDE3,用於心衰竭治療
磷脂酶 C / 肌醇磷酸鹽系統#
Gαq 活化磷脂酶 Cβ(PLCβ)→ 裂解膜磷脂 PIP₂ → 生成兩種第二傳訊者:
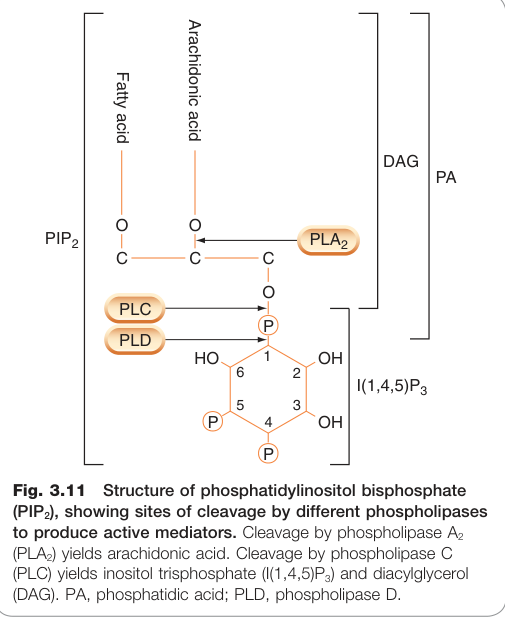
Figure 3.11:磷脂醯肌醇雙磷酸(PIP₂)結構,顯示各種磷脂酶的切割位點與活性介質的生成
- IP₃(肌醇 1,4,5-三磷酸):水溶性,釋放至細胞質,作用於內質網(ER)上的 IP₃ 受體(一種配體門控鈣通道),釋放 Ca²⁺ 至細胞質
- DAG(二醯甘油):脂溶性,留在膜上,活化蛋白激酶 C(PKC)→ 磷酸化多種細胞內蛋白質
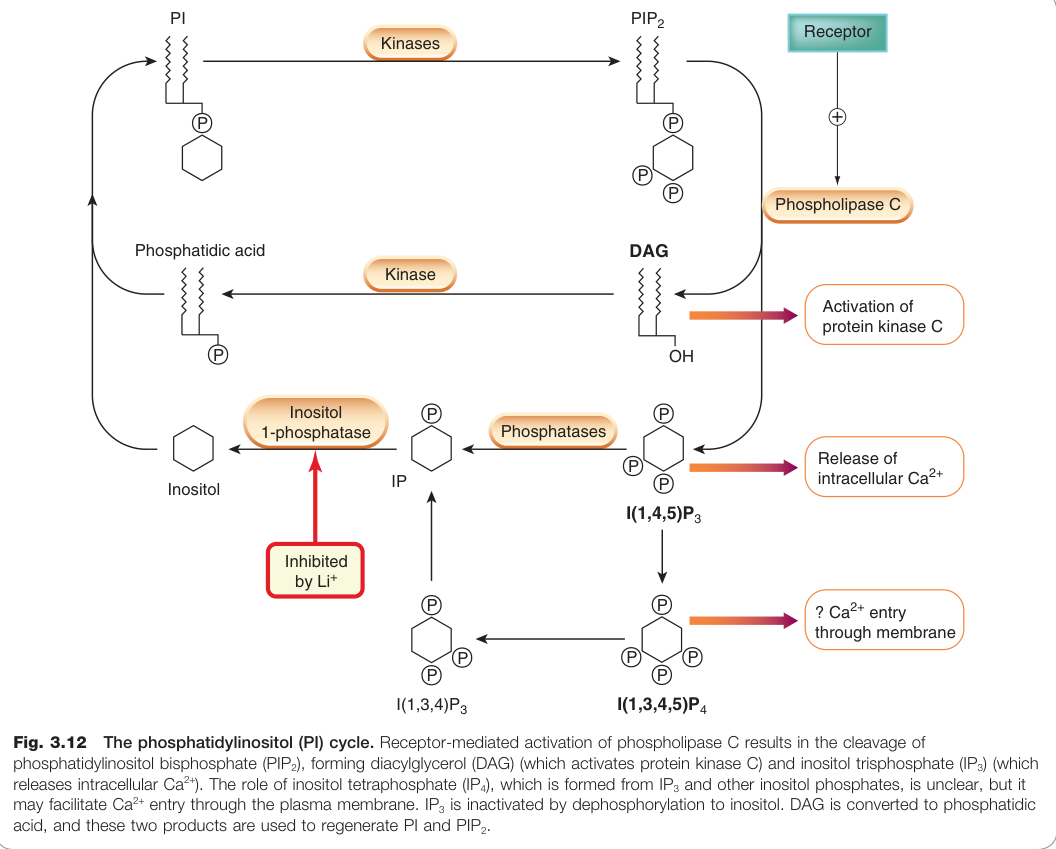
Figure 3.12:磷脂醯肌醇(PI)循環:受體活化磷脂酶 C 裂解 PIP₂,生成 DAG(活化 PKC)與 IP₃(觸發 Ca²⁺ 釋放)
鋰(lithium)阻斷 IP₃ 的脫磷酸酶循環路徑,這是其用於治療躁鬱症(bipolar disorder)的可能機制之一。
G 蛋白直接調控離子通道#
- Gβγ 次單元(尤其是 G₀ 型)可直接與鉀通道及鈣通道交互作用,無需第二傳訊者介入
- 例:心肌細胞毒蕈鹼型受體(mAChR)活化後,透過 G 蛋白直接增加 K⁺ 通透性,使細胞超極化
GPCR 去敏感化(Desensitisation)#
長期激動劑暴露會導致受體反應性降低:
- 同源去敏感化(homologous desensitisation):特異性 GPCR 激酶(GRKs)磷酸化已活化的受體 → 結合 β-arrestin → 受體無法與 G 蛋白結合 → 內吞(endocytosis)將受體移除出細胞膜
- 異源去敏感化(heterologous desensitisation):PKA 或 PKC 磷酸化多種受體,效果較弱且短暫
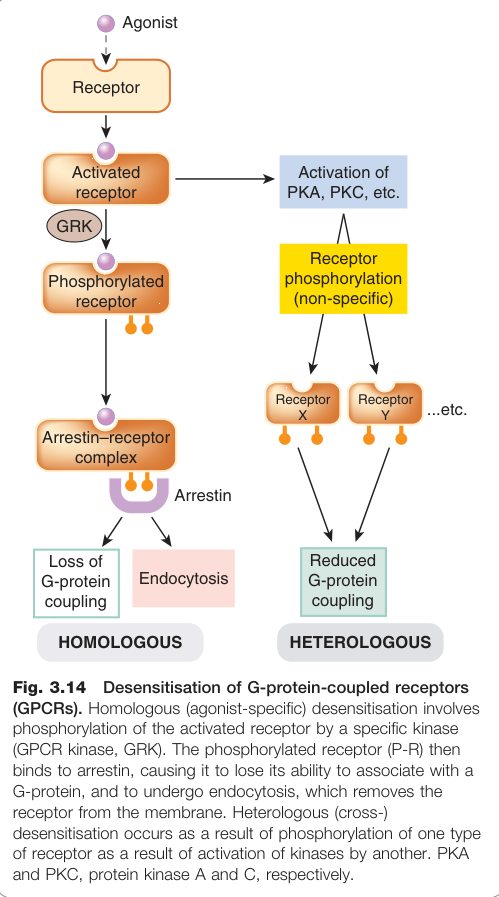
Figure 3.14:GPCR 去敏感化機制:同源去敏感化由 GRK 磷酸化活化受體並結合 β-arrestin 引發,異源去敏感化則由 PKA/PKC 非特異性磷酸化介導
近年研究顯示 β-arrestin 不只介導去敏感化,還可作為獨立的訊號傳遞接頭,連結 GPCR 至 MAP 激酶等路徑——這意味著不同激動劑可能在相同受體上引發質性不同的反應(稱為 biased agonism 或 agonist trafficking)。
GPCR 其他進展#
- GPCR 二聚化:許多 GPCR 以同源或異源二聚體形式存在,藥理特性不同於單體,如 GABA_B 受體需兩種亞型形成異二聚體才具功能
- RAMPs(受體活性調節蛋白):改變 GPCR 的配體特異性(如同一 GPCR 與 RAMP1 偶聯辨識 CGRP,與 RAMP2 偶聯則辨識腎上腺髓質素)
- RGS 蛋白(G 蛋白訊號調節蛋白):加速 Gα 次單元的 GTPase 活性,縮短 G 蛋白訊號持續時間
Type 3:激酶連結受體#
結構特徵#
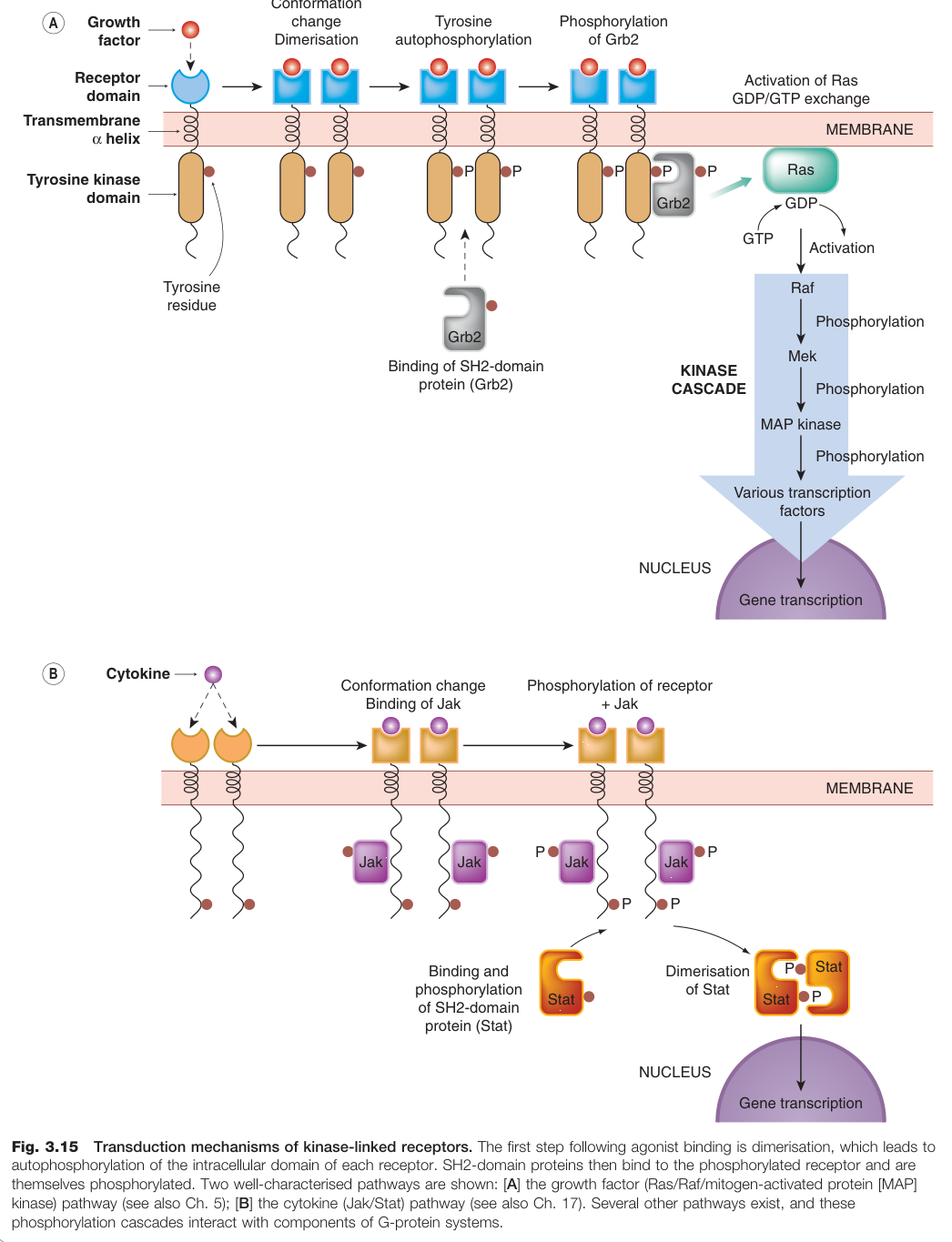
Figure 3.15:激酶連結受體的轉導機制:配體結合後受體二聚化,胞內域自我磷酸化,啟動下游訊號轉導
- 單一跨膜螺旋,連接大型胞外配體結合域與胞內激酶域
- 主要訊號轉導:配體結合 → 受體二聚化(dimerisation) → 胞內酪胺酸殘基自我磷酸化(autophosphorylation) → 磷酸酪胺酸作為 SH2 結構域蛋白的停靠位點 → 下游激酶串聯(kinase cascade)
主要類型#
- 受體酪胺酸激酶(RTKs):胞內域具有酪胺酸激酶活性,包括表皮生長因子受體(EGF receptor)、神經生長因子受體、胰島素受體(更複雜的二聚體結構)
- 絲胺酸/蘇胺酸激酶受體:如 TGF-β 受體
- 細胞激素受體(cytokine receptors):無內在酵素活性,活化後招募細胞質激酶(如 Janus kinase, Jak)
兩條重要下游路徑#
Ras/Raf/MAP 激酶路徑(生長因子訊號):
- RTK 自我磷酸化 → SH2 蛋白 Grb2 結合
- Ras(原癌基因產物)活化,進行 GDP/GTP 交換
- 依序活化 Raf → Mek → MAP 激酶
- MAP 激酶磷酸化轉錄因子 → 調控細胞分裂相關基因
Jak/Stat 路徑(細胞激素訊號):
- 細胞激素結合 → 受體二聚化 → Jak 激酶結合並磷酸化受體
- Stat 蛋白(SH2 結構域蛋白)結合磷酸化受體並被磷酸化
- 磷酸化 Stat 二聚化並進入核內 → 啟動基因轉錄
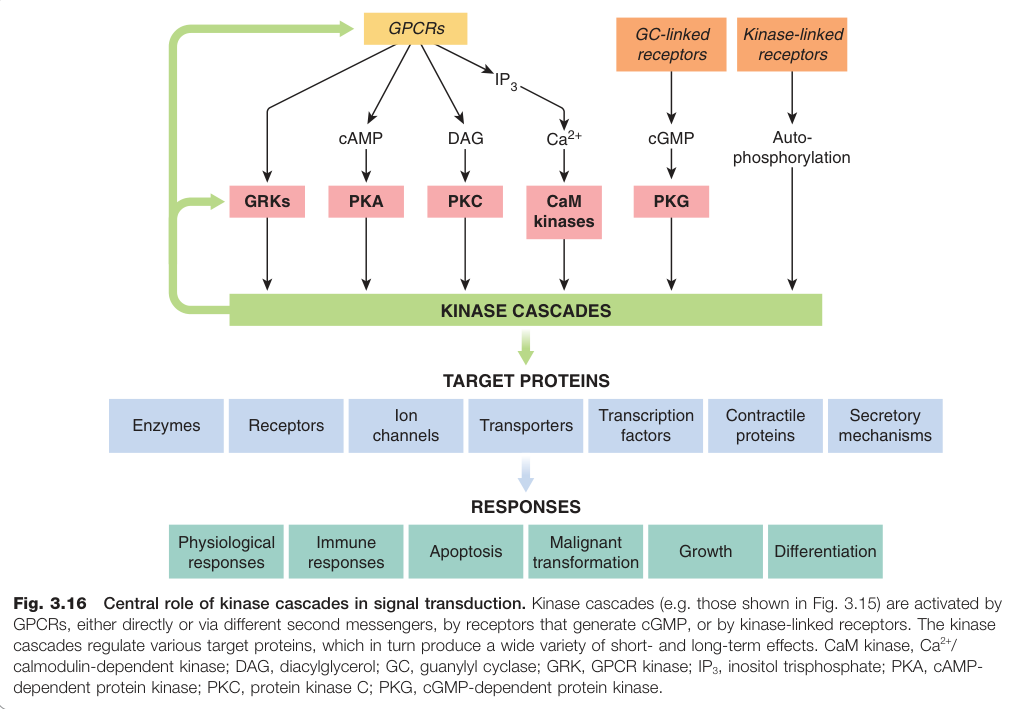
Figure 3.16:激酶串聯在訊號轉導中的核心角色:GPCR、受體酪胺酸激酶與細胞激素受體均可透過不同機制啟動激酶串聯
許多癌症與上述訊號路徑中的蛋白質基因突變有關,導致在無生長因子訊號的情況下路徑持續活化。第一個特異性激酶抑制劑 imatinib(基利克) 即針對慢性骨髓性白血病中一個特定酪胺酸激酶,開創了靶向激酶藥物治療的先河。
Type 4:核受體#
概述#
核受體(nuclear receptor, NR)家族由至少 48 個可溶性受體組成,以**配體活化轉錄因子(ligand-activated transcription factors)**的形式運作,透過調控基因轉錄介導細胞反應。
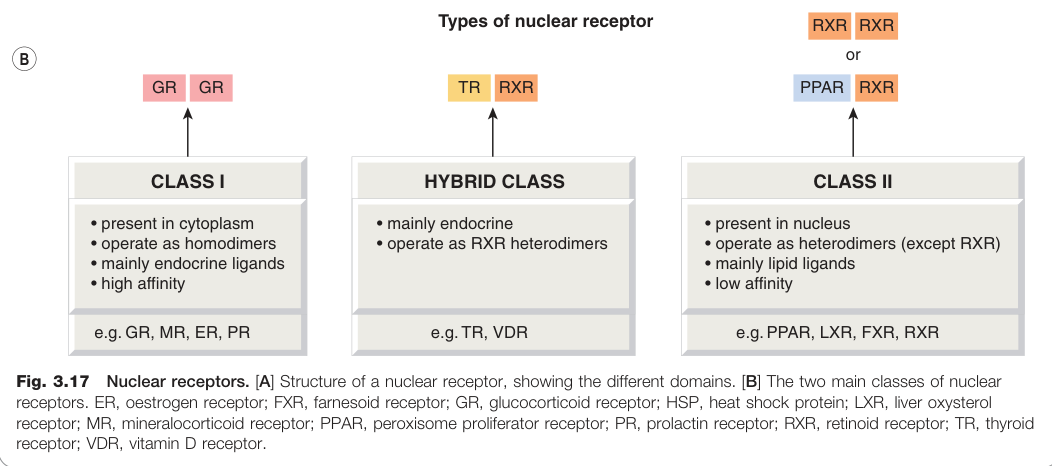
Figure 3.17:核受體結構與分類:[A] 核受體各功能域;[B] 兩大主要核受體類別(ER、FXR、GR 等代表成員)
- 配體通常是脂溶性小分子(類固醇荷爾蒙、甲狀腺素、視黃酸、維生素 D 等)
- 反應時間:數小時至數天(需蛋白質合成)
- 負責約 10% 處方藥的藥理作用;其調控的代謝酵素(如 CYP3A)影響約 60% 處方藥的藥物動力學
分子結構#
NR 共享相似的結構域:
- N 端 AF1 區域:配體非依賴性轉錄調節
- 核心 DNA 結合域:含「鋅指(zinc fingers)」,辨識荷爾蒙反應元素(hormone response elements)
- 鉸鏈區(hinge region):允許與其他 NR 形成二聚體
- C 端配體結合域:含 AF2 區域,配體依賴性活化
分類#
Class I(細胞質 NR):
- 無配體時在細胞質中與熱休克蛋白(HSP)結合
- 配體結合後形成同源二聚體(homodimer),轉位至核內
- 代表例:糖皮質素受體(GR)、礦皮質素受體(MR)、雌激素受體(ER)、孕酮受體(PR)
Class II(核內 NR):
- 幾乎都以**異源二聚體(heterodimer)**形式與 RXR(retinoid X receptor)運作
- 配體通常是脂質類物質(脂肪酸、膽固醇衍生物等)
- 代表例:PPAR(過氧化物酶增殖物活化受體)、LXR(肝臟氧甾醇受體)、FXR(膽汁酸受體)
SXR/PXR 與 CAR 等 Class II NR 是重要的「外源物感知器」,能辨識許多外源化合物(包括治療藥物)並誘導 CYP3A 等藥物代謝酵素,是藥物交互作用的重要機制。
第三亞群(甲狀腺素型):形成與 RXR 的異源二聚體,但主要傳遞內分泌訊號,包括甲狀腺素受體(TR)、維生素 D 受體(VDR)和視黃酸受體(RAR)。
基因轉錄調控#
- NR 結合至基因啟動子區的荷爾蒙反應元素(短 4–5 個鹼基對序列)
- 招募共活化因子(co-activators):包括組蛋白乙醯化酶(histone acetylase),使染色質鬆解,促進基因轉錄
- 招募共抑制因子(co-repressors):包括組蛋白去乙醯化酶(histone deacetylase),使染色質緊密,抑制轉錄
離子通道作為藥物靶點#
除配體門控離子通道外,其他類型的離子通道也是重要藥物靶點。
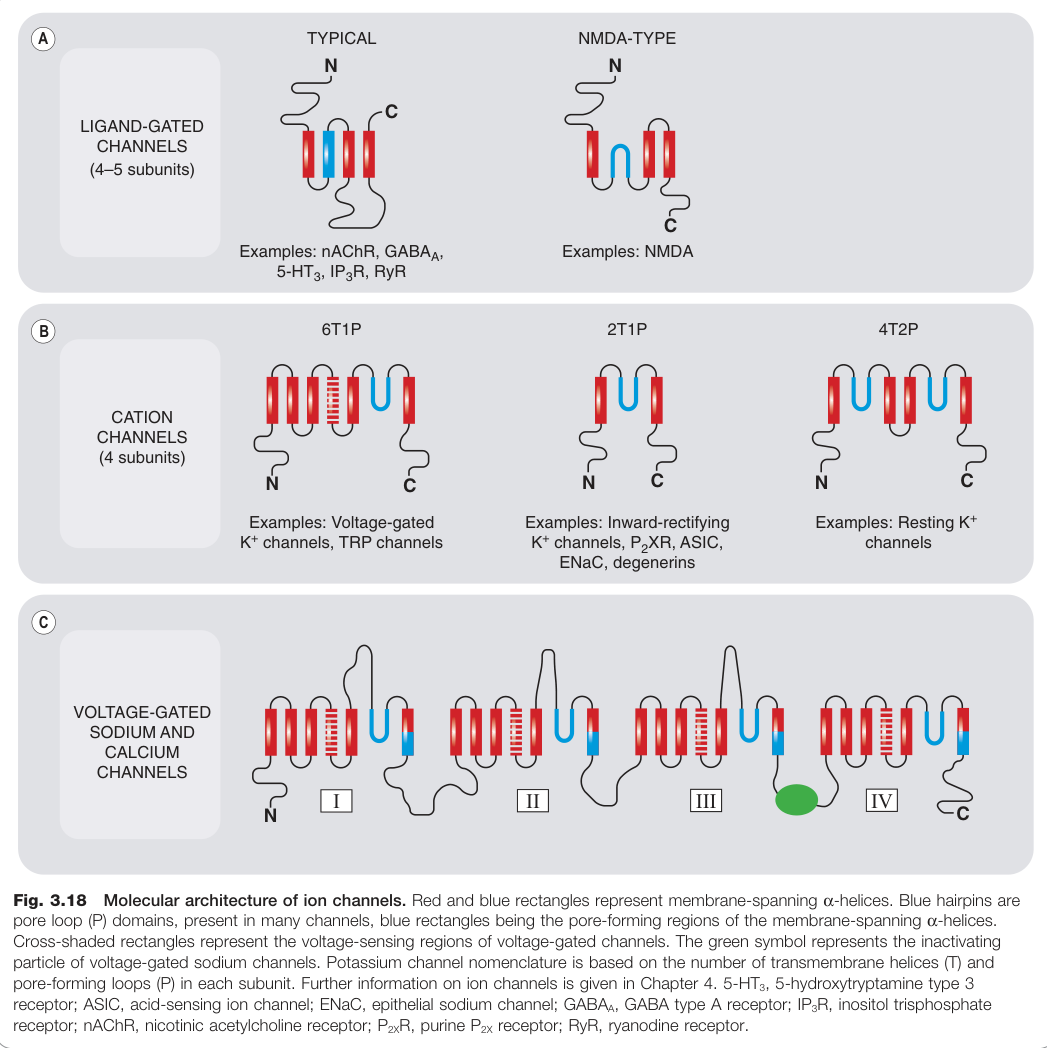
Figure 3.18:離子通道的分子架構:紅、藍矩形代表跨膜 α-螺旋,藍色髮夾為 pore loop(P)結構域
離子選擇性#
- 陽離子選擇性通道:Na⁺ 、Ca²⁺ 或 K⁺ 選擇性,或對三者均通透
- 陰離子通道:主要通透 Cl⁻
閘控類型#
- 電位門控通道(voltage-gated channels):膜去極化時開放,包括 Na⁺、K⁺、Ca²⁺ 通道;部分在活化後會發生「失活(inactivation)」以自限
- 配體門控通道(見 Type 1 受體)
- 胞內訊號門控:
- Ca²⁺ 活化鉀通道:[Ca²⁺]ᵢ 升高時開放,使細胞超極化
- ATP 敏感性鉀通道(K_ATP):細胞 ATP 耗竭時開放(胰臟 β 細胞的血糖偵測機制中關鍵)
- 鈣釋放通道:位於內質網,包括 IP₃ 受體和 ryanodine 受體(RyR)
- 儲存操控鈣通道(store-operated Ca²⁺ channels, SOCs):胞內 Ca²⁺ 儲存耗竭時開放,維持 Ca²⁺ 訊號並補充儲存
藥物影響離子通道的機制#
- 通道阻斷:如河豚毒素(tetrodotoxin)、局部麻醉藥物阻斷電位門控鈉通道
- 失活阻斷(block of inactivation):如韋拉曲林(veratridine)、烏頭毒素(batrachotoxin)
- 間接調控(經 GPCR 和第二傳訊者磷酸化通道蛋白):如 β 腎上腺素受體激動劑調控鈣通道和鉀通道
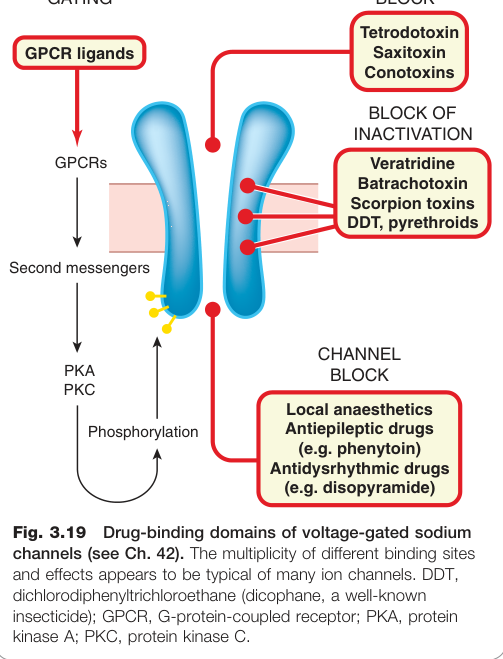
Figure 3.19:電位門控鈉通道的多重藥物結合位點,DDT 等多種化合物作用於不同位點,顯示離子通道藥理複雜性
受體表現的調控#
受體蛋白並非固定不變,其表現量本身受到調控:
- 短期調控:去敏感化(受體磷酸化與內吞)
- 長期調控:受體表現上調或下調
- 去神經後突觸後受體增生(upregulation)
- 炎症時多種 GPCR 和細胞激素受體表現增加
- 腫瘤病毒誘導生長因子受體表現
- 長期藥物治療導致適應性反應(adaptive response),可能是抗憂鬱藥起效緩慢或藥物依賴性的基礎
受體與疾病#
受體功能異常可直接導致多種疾病,主要機制:
- 自體抗體對受體蛋白的攻擊:如重症肌無力(myasthenia gravis)——自體抗體使 nAChR 失活;Graves 病——活化抗體模擬 TSH 促使甲狀腺持續分泌
- 編碼受體或訊號蛋白的基因突變:
- 促進性突變(gain-of-function):如促甲狀腺素受體突變導致甲亢、黃體素受體突變導致性早熟
- 喪失性突變(loss-of-function):如抗利尿素受體突變導致對荷爾蒙抵抗
β₂-腎上腺素受體的多型性(polymorphism)雖不直接致病,但與氣喘患者對 β 腎上腺素受體激動劑治療效果較差、心衰竭預後不佳有關。這是藥物基因體學(pharmacogenomics)的重要研究領域。