概述#
藥理學作為一門科學的發展,始於研究重心從「描述藥物做什麼」轉向「解釋藥物如何作用」。本章建立藥物與生命系統交互作用的一般原則,後續第三章將深入探討分子層面的機制。
Paul Ehrlich(保羅·埃利希)奠定了現代藥理學的基礎——他堅持藥物作用必須以藥物與組織之間的化學交互作用來解釋,並提出著名的格言:Corpora non agunt nisi fixata(「藥物不結合則不起效」)。
藥物作用標靶#
藥物必須與細胞成分發生化學作用才能產生藥理反應,其前提是藥物分子必須「結合」到特定細胞成分上,呈非均勻分佈。這些關鍵結合位點通稱為藥物標靶(drug targets)。
絕大多數藥物標靶是蛋白質分子,可分為四大類:
- 受體(receptors)
- 酵素(enzymes)
- 載體蛋白(carrier molecules / transporters)
- 離子通道(ion channels)
少數例外包括:部分抗菌藥與抗腫瘤藥直接與 DNA 作用;雙磷酸鹽類藥物(biphosphonates)則與骨基質中的鈣鹽結合。此外,滲透性利尿劑、制酸劑等藥物幾乎不需結合組織即可發揮作用,但這些例外不影響上述原則的普遍適用性。
藥物特異性#
特異性是雙向的:
- 個別藥物只結合特定類型的標靶
- 個別標靶也只辨識特定類型的藥物
然而,沒有任何藥物具有完全的特異性。劑量越高、效力越低,越可能同時作用於非主要標靶,導致副作用。例如三環類抗憂鬱藥(tricyclic antidepressants)阻斷單胺轉運體的同時,也會阻斷多種受體而引起口乾等副作用。
受體的概念#
受體在生理系統中的角色#
受體(receptor)是多細胞生物協調各器官細胞活動的化學通訊系統中的核心元件。
以腎上腺素(adrenaline / epinephrine)對心臟的作用為例:
- 腎上腺素結合到 β-腎上腺素受體(β-adrenoceptor)
- 結合後觸發一系列反應,增加心臟的收縮力與心率
- 若無腎上腺素,受體功能上沉默
促效劑(agonist) 與 拮抗劑(antagonist) 的核心區別:
- 促效劑:結合受體並「啟動」受體
- 拮抗劑:結合相同位點但不引起啟動,因而阻斷促效劑的效果
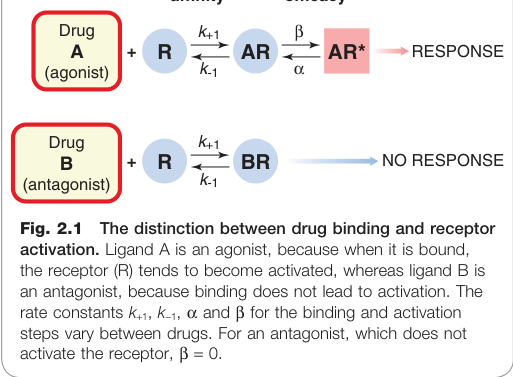
Figure 2.1:藥物結合與受體活化的區別——配體 A 為促效劑,配體 B 為拮抗劑
受體分類#
受體分類是藥物設計的重要工具。以組織胺(histamine)為例,Black 等人於 1970 年提出 H2 受體的假說,進而開發出選擇性 H2 拮抗劑,對於治療胃酸分泌具有重大臨床意義。
受體分類的依據包括:
- 藥理學反應(傳統方法)
- 放射性配體結合研究(直接測量親和力)
- 分子選殖(molecular cloning)(提供最精細的分類層級)
- 訊號傳遞路徑分析
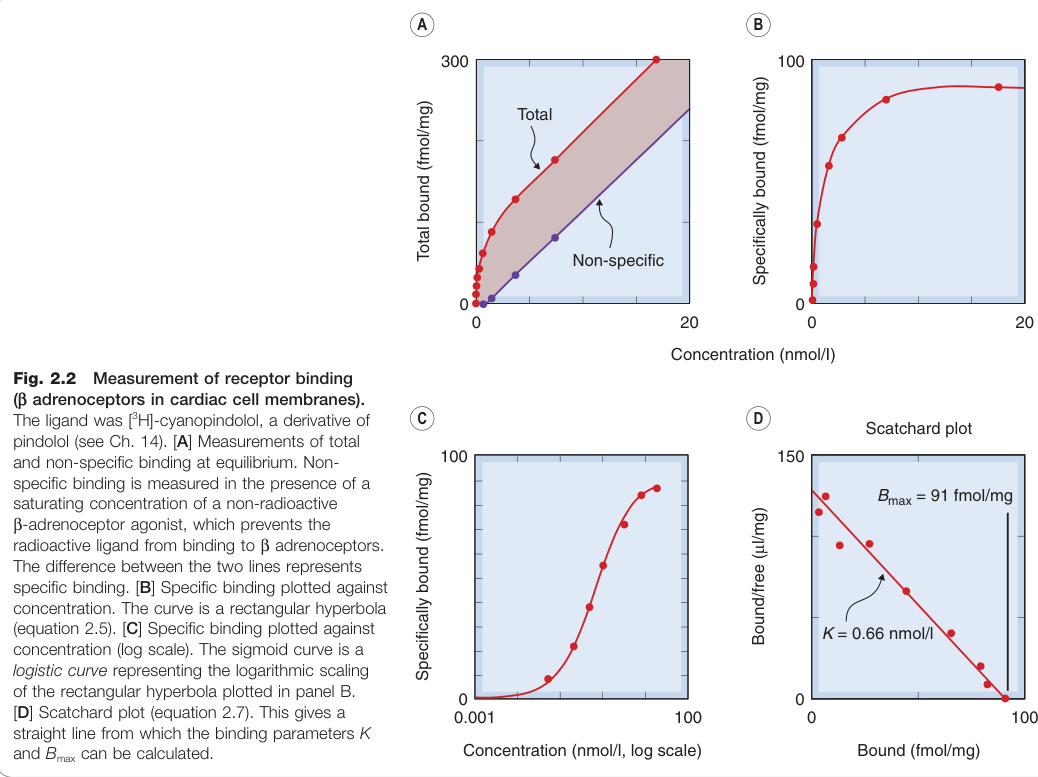
Figure 2.2:以放射性配體 [³H]-cyanopindolol 測量心肌細胞膜 β 腎上腺素受體結合量
國際藥理科學聯合會(IUPHAR)設有受體命名委員會,負責整合藥理、分子與生化資訊,制定統一的受體分類術語。
藥物–受體交互作用#
結合(Binding)與啟動(Activation)#
藥物佔據受體與受體被啟動是兩個獨立步驟(見圖 2.1 概念):
- 藥物與受體結合的傾向,由其**親和力(affinity)**決定
- 藥物結合後啟動受體的傾向,由其**效能(efficacy)**決定
| 藥物類型 | 親和力 | 效能 |
|---|---|---|
| 完全促效劑(full agonist) | 高 | 高 |
| 部分促效劑(partial agonist) | 有 | 中等 |
| 拮抗劑(antagonist) | 有 | 零 |
| 反向促效劑(inverse agonist) | 有 | 負值 |
藥物濃度–效應曲線#
生物反應常以**濃度–效應曲線(concentration–effect curve)或劑量–反應曲線(dose–response curve)**呈現,可得到:
- Emax:藥物能產生的最大反應
- EC₅₀ / ED₅₀:產生 50% 最大反應所需的濃度或劑量
濃度–效應曲線的形狀與受體結合曲線相似,但不能直接用來測定促效劑對受體的親和力,因為生理反應通常與受體佔據率不成正比關係。此外,組織中的藥物濃度(如乙醯膽鹼被膽鹼酯酶分解後)可能遠低於浴液中的濃度。
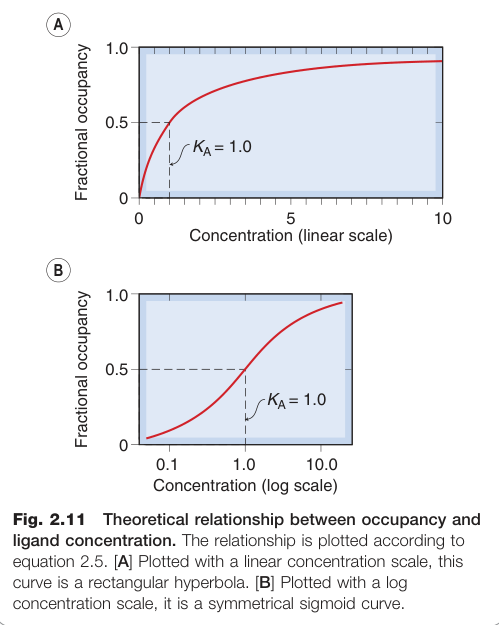
Figure 2.11:佔據率與配體濃度的理論關係——以線性及對數濃度軸分別繪製的雙曲線與 S 型曲線
競爭性拮抗(Competitive Antagonism)#
競爭性拮抗是最重要的拮抗機制,具有以下特徵:
- 促效劑對數濃度–效應曲線向右平移,斜率與最大值不變
- 劑量比(dose ratio, r)與拮抗劑濃度呈線性關係
- 此阻斷為可克服的(surmountable):提高促效劑濃度可恢復反應
可逆性競爭拮抗中,拮抗劑解離速率夠快,促效劑可置換受體上的拮抗劑。不可逆性競爭拮抗則是拮抗劑以共價鍵與受體結合(如阿斯匹靈對 COX 酵素的不可逆抑制)。
拮抗劑親和力(KB)可由 Schild 方程式(Schild equation)計算,對應的效力指標稱為 pA₂(使劑量比等於 2 所需拮抗劑濃度的負對數),廣泛用於受體分類。
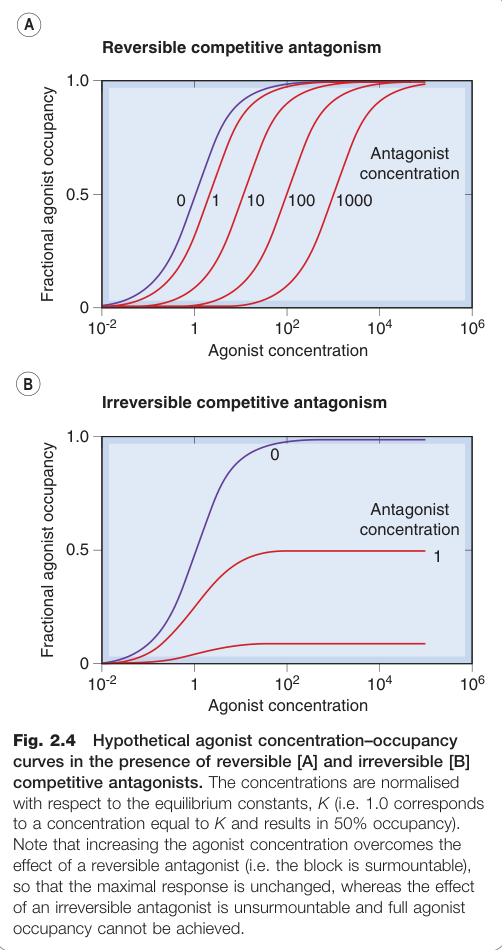
Figure 2.4:可逆性與不可逆性競爭拮抗劑存在下,假設性促效劑濃度–佔據率曲線的右移情形
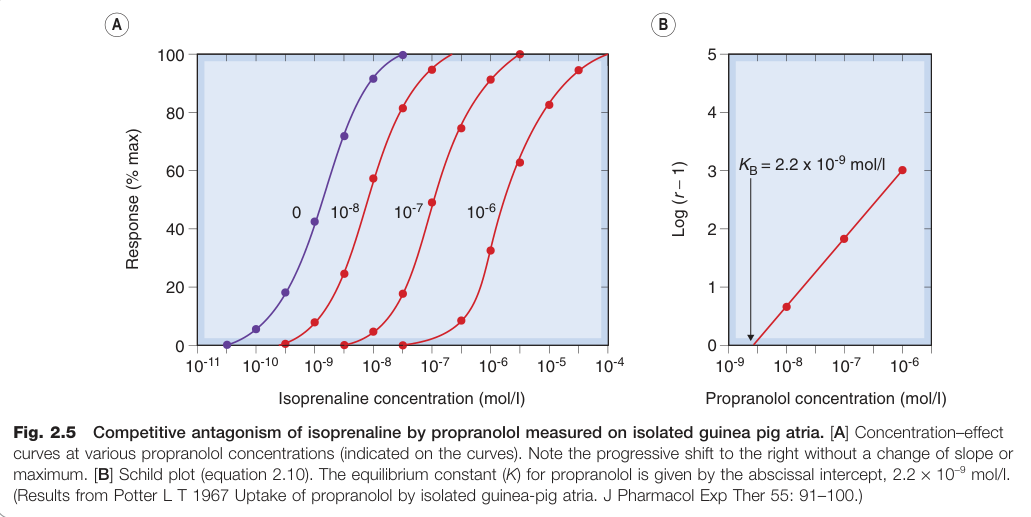
Figure 2.5:普萘洛爾(propranolol)對異丙腎上腺素(isoprenaline)的競爭性拮抗——以離體天竺鼠心房測量
變構效應(Allosteric Effects)#
除了促效劑結合位點外,受體蛋白還具有其他**變構(allosteric)**結合位點,藥物可透過這些位點:
- 增加或降低促效劑的親和力
- 修改效能
典型例子:苯二氮平類藥物(benzodiazepines)對 GABA_A 受體的變構促進作用;甘胺酸(glycine)對麩胺酸受體(glutamate receptors)的變構調節。
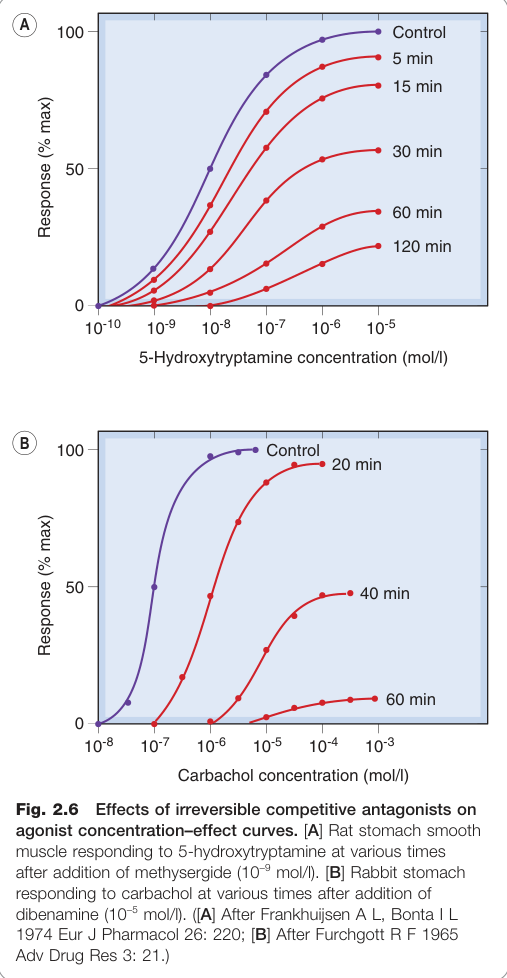
Figure 2.6:不可逆性競爭拮抗劑對促效劑濃度–效應曲線的影響,包括 5-HT 在胃平滑肌的實驗結果
部分促效劑與效能#
效能的定義#
當同一類型受體上的不同促效劑被測試時,即使在高濃度下,各藥物能引起的最大反應也可能不同:
- 完全促效劑(full agonist):能產生組織最大反應
- 部分促效劑(partial agonist):只能產生次最大反應(submaximal response)
效能(efficacy, e)最初由 Stephenson(1956)定義,描述藥物–受體複合物引發組織反應的「強度」。在雙態模型中,效能反映藥物–受體複合物趨向活化態(AR*)而非靜止態(AR)的傾向。
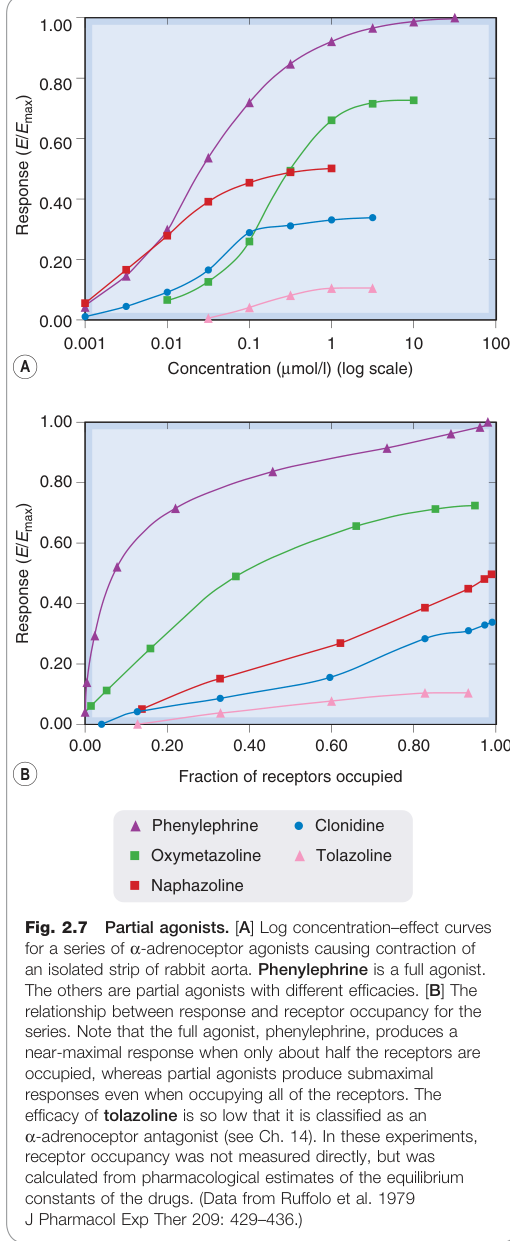
Figure 2.7:部分促效劑——系列 α 腎上腺素受體促效劑對離體兔主動脈的對數濃度–效應曲線,苯腎上腺素(phenylephrine)為完全促效劑
腎上腺素(adrenaline)與普萘洛爾(propranolol)對 β-腎上腺素受體的親和力相近,但效能截然不同——前者為促效劑,後者為拮抗劑。混淆兩者在臨床上後果嚴重。
雙態受體模型(Two-State Receptor Model)#
受體存在於「靜止態(R)」和「活化態(R*)」的動態平衡中:
- 促效劑:對 R* 的親和力大於 R,推動平衡向 R* 移動
- 完全促效劑:對 R* 的選擇性極高,幾乎所有佔據的受體均採用 R* 構型
- 部分促效劑:對 R* 的選擇性中等
- 中性拮抗劑(neutral antagonist):對 R 與 R* 親和力相同,不影響平衡
- 反向促效劑(inverse agonist):對 R 的親和力大於 R*,將平衡推向靜止態
雙態模型是有用的起點,但過於簡化——受體實際上具有多種構型狀態,可啟動不同的訊號傳遞路徑(「偏向性促效(biased agonism)」的概念)。
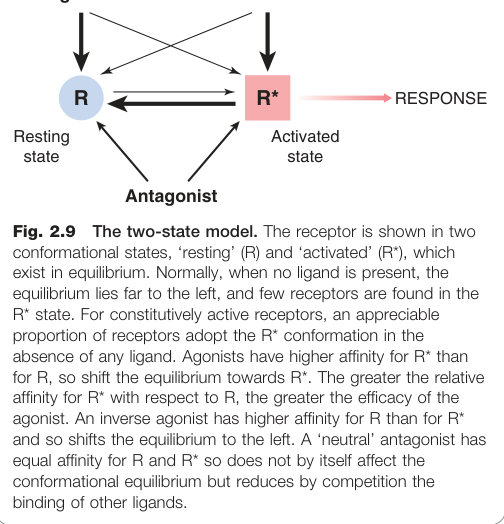
Figure 2.9:雙態模型——受體在「靜止態(R)」與「活化態(R*)」之間的平衡,及不同配體對平衡方向的影響
構成性受體活化與反向促效劑#
某些受體即使無配體存在,也會有相當程度的自發活化(構成性活化,constitutive activation),例如:
- 苯二氮平受體
- 大麻素受體(cannabinoid receptors)
- 血清素受體(serotonin receptors)
在這種情況下,反向促效劑(inverse agonist)能降低基礎活化水準,而中性拮抗劑則不影響基礎活化水準,僅競爭性阻斷促效劑的效果。
反向促效劑在理論上比中性拮抗劑更適合治療與構成性受體活化相關的疾病(如某些甲狀腺功能亢進症、性早熟),但目前多數用於臨床的拮抗劑事實上是反向促效劑。
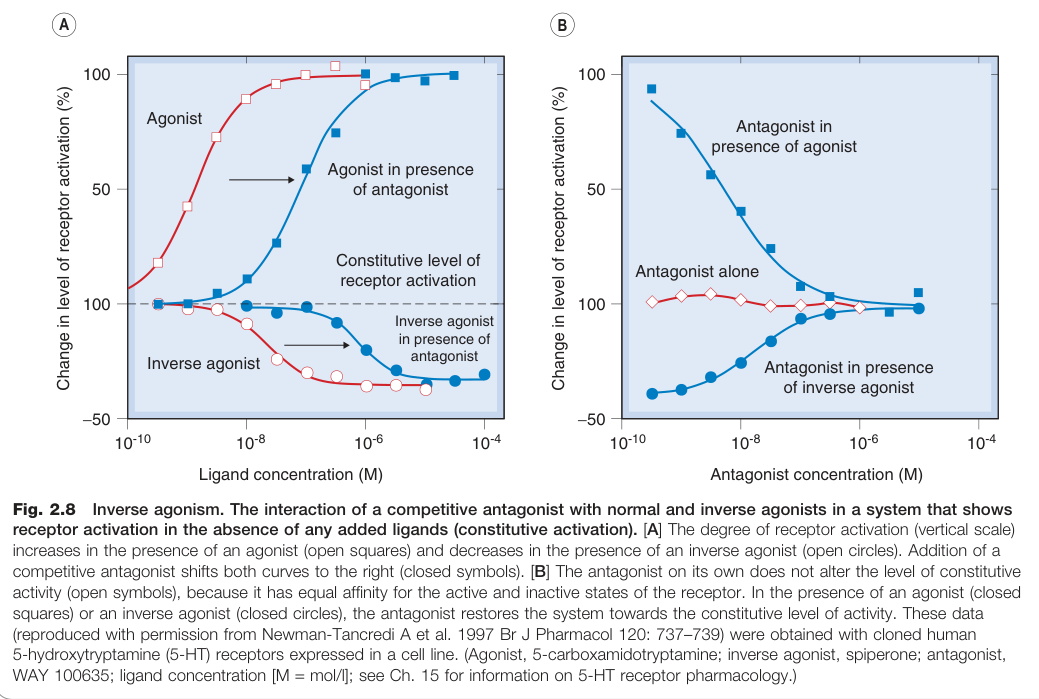
Figure 2.8:反向促效劑——在具有構成性活化受體的系統中,競爭性拮抗劑與正向及反向促效劑的交互作用
備用受體(Spare Receptors)#
Stephenson 等人觀察到,許多完全促效劑在受體佔據率極低(常低於 1%)時即可產生最大反應,表示系統中存在「備用受體」(spare receptors)或「受體儲備(receptor reserve)」。
意義:
- 並非受體在功能上有所分組,而是受體總數超過引發完整反應所需的數量
- 使激素或神經遞質在較低濃度時即可達到最大效應,節省分泌資源
藥物拮抗的機制#
藥物拮抗可透過多種機制發生:
- 化學拮抗(chemical antagonism):兩藥在溶液中直接結合(如螯合劑二巰基丙醇 dimercaprol 與重金屬結合)
- 藥代動力學拮抗(pharmacokinetic antagonism):一藥影響另一藥的吸收、代謝或排泄(如苯巴比妥加速華法林的肝臟代謝)
- 競爭性拮抗(competitive antagonism):兩藥競爭同一受體
- 受體–效應連結阻斷(block of receptor–effector linkage):在受體下游阻斷訊號傳遞(如鈣通道阻斷劑維拉帕米 verapamil 阻斷平滑肌收縮)
- 生理性拮抗(physiological antagonism):兩藥產生相反的生理效應(如組織胺刺激胃酸分泌,奧美拉唑 omeprazole 抑制質子泵)
脫敏與快速耐受性#
持續或反覆給藥時,藥物效應往往逐漸減弱,相關術語如下:
- 脫敏(desensitisation)/ 快速耐受性(tachyphylaxis):數分鐘內發展
- 耐受性(tolerance):數天至數週緩慢發展
- 抗藥性(drug resistance):主要指抗菌或抗腫瘤藥的失效
主要機制#
- 受體變化(change in receptors):
- 離子通道型受體:促效劑結合後受體發生構型改變,離子通道關閉
- G 蛋白偶聯受體(G-protein-coupled receptors):磷酸化導致受體與 G 蛋白「脫聯(uncoupling)」
- 受體內化(translocation / internalisation):受體從細胞表面減少(如 β-腎上腺素受體在異丙腎上腺素存在下,8 小時內密度可降至正常的 10%)
- 介質耗竭(exhaustion of mediators):如安非他命(amphetamine)持續刺激胺類釋放導致儲存耗盡
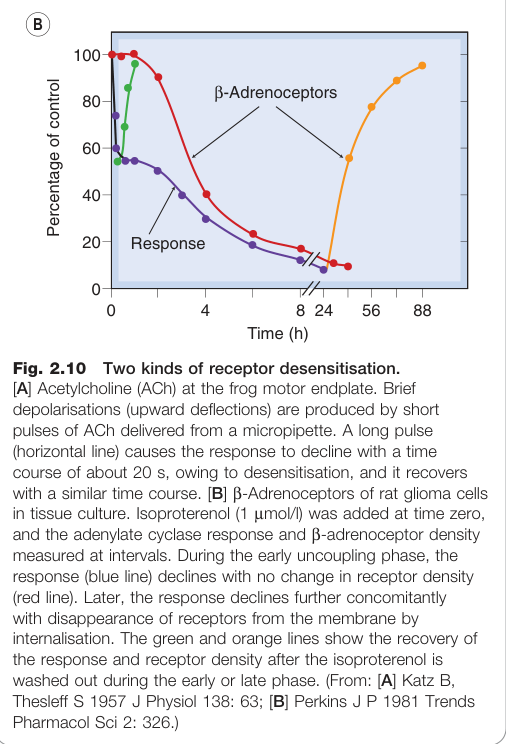
Figure 2.10:兩種受體脫敏類型——ACh 在蛙類運動終板的快速離子通道型脫敏,及 β 腎上腺素受體的慢速 G 蛋白偶聯型脫敏
- 代謝加速(altered drug metabolism):如巴比妥類、乙醇誘導自身的代謝酵素
- 生理性適應(physiological adaptation):如降壓藥引發代償性腎素–血管張力素系統活化
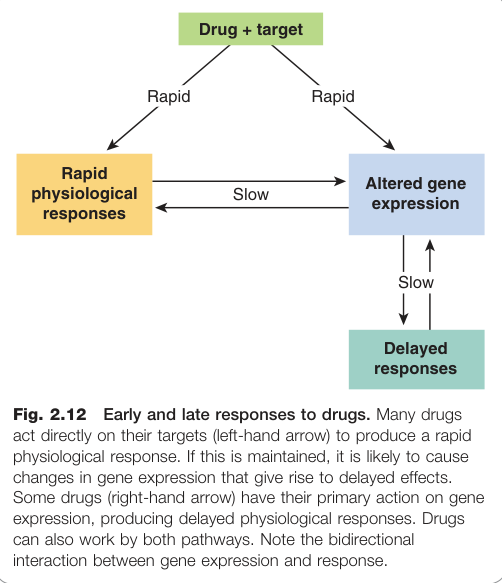
藥物效應的時間層次#
藥物的直接效應僅是第一步,後續衍生的延遲效應在臨床上同樣重要。
藥物效應在時間軸上呈現多個層次:
- 快速反應(秒至分鐘):直接的生化或生理反應(如支氣管擴張劑立即緩解哮喘)
- 中等速度(分鐘至小時):脫敏、受體下調(down-regulation)
- 延遲反應(小時至天):基因表現改變引起的結構重塑(如心臟肥大)
典型例子:
- 抗憂鬱藥立即影響神經傳遞物代謝,但需數週才出現療效
- 鴉片類藥物(opioids)立即止痛,但長期使用後發展出耐受性與依賴性
任何長期的表現型改變都必然涉及基因表現的改變,這使得長期藥物效應的研究愈發重要。