正常止血(hemostasis)需要凝血因子(coagulation factors)與血小板(platelets)共同協調運作。任一環節缺陷都可能造成出血傾向。本章涵蓋血友病(hemophilia)、von Willebrand 病(von Willebrand disease, vWD)、其他凝血因子缺乏、免疫性血小板低下症(immune thrombocytopenia, ITP)與血栓性微血管病變(thrombotic microangiopathies, TMA)。
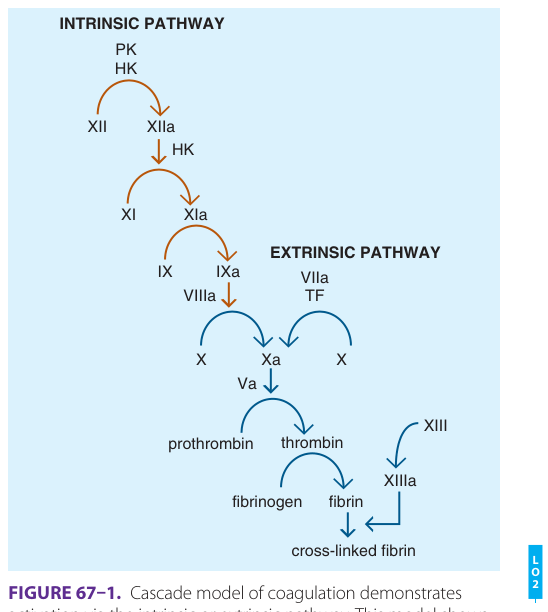
Figure 67–1:凝血的瀑布模型
血友病#
流行病學與分類#
血友病為 X 染色體性聯遺傳,主要影響男性。
- 血友病 A(hemophilia A):第八凝血因子(factor VIII)缺乏。
- 血友病 B(hemophilia B):第九凝血因子(factor IX)缺乏。
兩型臨床表現無法區分,皆依因子活性分為重度(< 1%)、中度(1%–5%)、輕度(> 5%)。隨著有效治療進展,病人平均壽命已接近一般人口(約 63 至 75 歲)。
臨床表現與診斷#
- 關節腔出血(hemarthrosis)為血友病的標誌,好發於膝、肘、踝關節,反覆未妥善處理會導致血友病性關節病變(hemophilic arthropathy)與慢性疼痛。
- 其他表現:瘀斑、肌肉出血、口腔與泌尿生殖道出血、消化道出血、顱內出血、手術時過度出血。
- 實驗室檢查:凝血酶原時間(prothrombin time, PT)正常、血小板正常、活化部分凝血活酶時間(activated partial thromboplastin time, aPTT)延長;血友病 A 為 factor VIII 低下、血友病 B 為 factor IX 低下。
治療目標#
- 短期目標:減少每年出血次數與出血頻率、使凝血因子濃度正常化或改善。
- 長期目標:維持關節功能、使骨科與放射學關節評分正常化、維持生活品質。
治療#
因子補充與預防
靜脈注射重組型(recombinant)或血漿衍生(plasma-derived)凝血因子製劑以治療或預防出血,是血友病治療的核心。初級預防(primary prophylaxis)指在尚未出現關節病變、且 3 歲前即開始規律給予因子製劑,建議用於重度因子缺乏者,可有效預防出血及相關併發症。
血友病 A — DDAVP:1-desamino-8-D-arginine vasopressin(desmopressin acetate,DDAVP)為血管加壓素類似物,可促進內皮儲存的 von Willebrand factor(vWF)與 factor VIII 釋放。建議劑量 0.3 mcg/kg 靜脈輸注或皮下注射,或濃縮鼻噴劑 150–300 mcg。適用於輕度血友病 A 的輕微出血,不可用於嚴重危及生命的出血,因反應延遲且 factor VIII 上升幅度可能不足。需限制飲水以防低血鈉(hyponatremia),2 歲以下不建議使用。
抗纖維蛋白溶解治療:aminocaproic acid 與 tranexamic acid 抑制纖溶酶活性、穩定血塊,多作為牙科處置或經血過多的輔助治療,須與適當因子製劑併用。
factor VIII 補充:依出血嚴重度與位置決定矯正目標與療程。輕微出血目標為正常活性的 25%–30%,嚴重或危及生命出血需 > 80%。每 1 unit/kg factor VIII 約使活性上升 2%。劑量計算:
factor VIII 劑量(units)= 體重(kg)×(欲提升的百分比)× 0.5
factor VIII 半衰期約 9 至 10 小時,每半衰期(每 8–12 小時)給予初始劑量的一半以維持目標濃度。長時間治療可採連續輸注以避免危險的低谷濃度。
血友病 B — factor IX 補充:因 factor IX 分子較小、分布於血管內外,每 1 unit/kg 僅使活性上升約 1%。劑量計算需乘以校正因子 F(血漿衍生產品 F = 1;Benefix 成人 F = 1.4、兒童 1.2)。半衰期約 16 至 17 小時,每 18 至 24 小時給藥。較舊的凝血酶原複合物濃縮劑(prothrombin complex concentrates, PCCs)因含其他維生素 K 依賴蛋白具血栓風險,並非首選。
factor IX 劑量(units)= 體重(kg)×(欲提升的百分比)× F
因子抑制物的處置
約 30% 的血友病 A 與 5% 的血友病 B 病人會對補充治療產生中和性抗體(抑制物)。抑制物效價以 Bethesda 單位(BU)量測以指導治療。
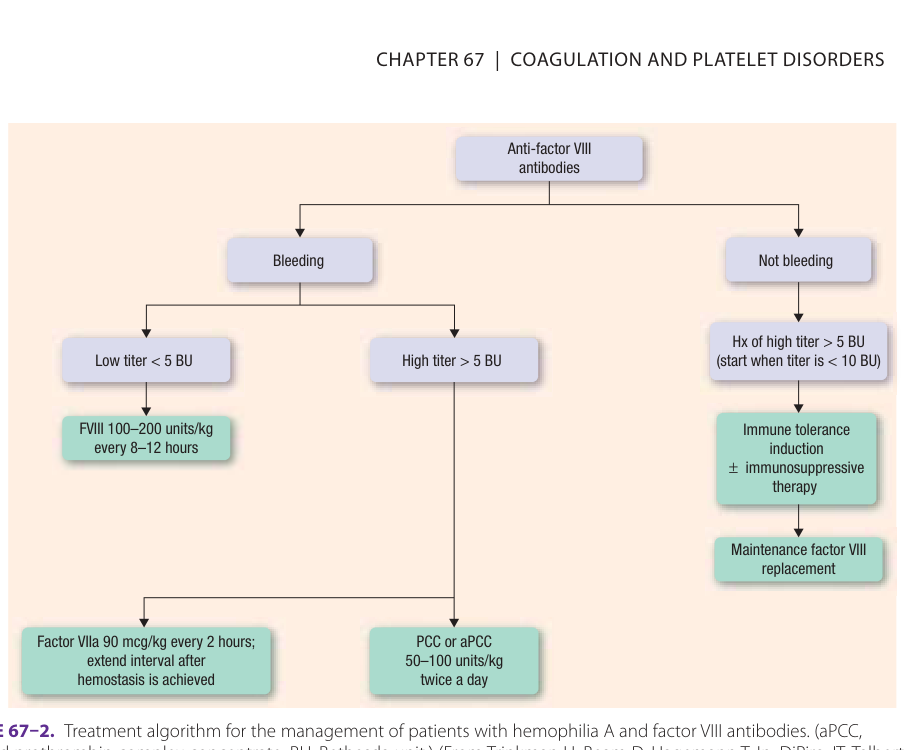
Figure 67–2:血友病 A 合併第八因子抗體的處置流程
- 低效價(< 5 BU):可加大 factor VIII 劑量控制急性出血。
- 高效價:使用繞道製劑(bypassing agents),包括 PCCs、活化型 PCC(aPCC,FEIBA)與重組型 factor VIIa(rFVIIa)。rFVIIa 僅在組織損傷部位產生凝血酶,全身性血栓事件較 PCC 少。recombinant porcine factor VIII(Obizur)為第三線。
- 免疫耐受誘導(immune tolerance induction)可消除抑制物,對血友病 A 有效率約 70%、血友病 B 約 30%。
疼痛處置
acetaminophen 與 opioid 分別用於輕中度與重度疼痛。應避免 NSAID 與 aspirin,因其與血小板結合而增加出血風險;COX-2 抑制劑可謹慎使用。
von Willebrand 病(vWD)#
流行病學與病理生理#
vWD 是最常見的遺傳性出血疾病,盛行率約 0.6%–1.3%,多為體染色體顯性遺傳,男女比例相當。vWF 為大型多聚體醣蛋白,在止血中協助血小板黏附受損血管壁,並攜帶與穩定血漿中的 factor VIII。
vWD 依缺陷分型:
- 第 1 型:vWF 與 factor VIII 部分量性缺乏(佔 70%–80%),體染色體顯性。
- 第 2 型:vWF 質性異常(2A、2B、2M、2N 亞型)。
- 第 3 型:vWF 嚴重量性缺乏,體染色體隱性,罕見。
臨床表現包括瘀青、黏膜皮膚出血、鼻出血、口腔出血、經血過多、消化道出血與術後出血;輕症者可能至成年仍無症狀。
治療#
vWD 出血傾向通常較血友病少且輕,多不需慢性預防。目標為視需要止住自發性出血、預防手術與產後出血。兩大全身性治療為刺激內生性 vWF 釋放的 DDAVP,或補充含 vWF 的製劑。
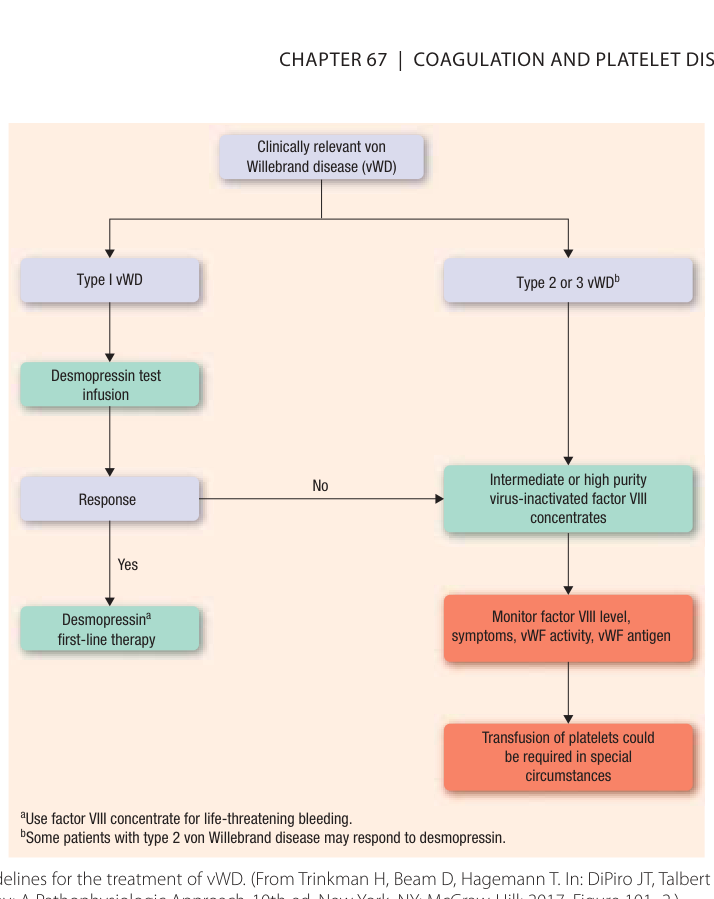
Figure 67–3:von Willebrand 病的治療指引
- DDAVP:第 1 型且功能正常 vWF 者的首選,劑量同血友病 A。可使 vWF 與 factor VIII 升高 2 至 5 倍,效果優於血友病。對分泌異常 vWF 的 2A 型通常無效;2B 型可能增加血小板低下風險。第 3 型缺乏可釋放的 vWF,對 DDAVP 無反應。
- 抗纖溶治療:aminocaproic acid 與 tranexamic acid 用於鼻出血與經血過多。
- 補充治療:對 DDAVP 無反應的第 1 型、第 2 及 3 型、與大手術病人,給予含 vWF 的中至高純度病毒去活化 factor VIII 濃縮劑。超高純度(單株)與重組型 factor VIII 不含 vWF,不應用於 vWD。
其他凝血因子缺乏#
隱性遺傳凝血障礙(recessively inherited coagulation disorders, RICDs)指 factor II、V、VII、X 至 XIII 等相對罕見的缺乏,造成因子產量減少或功能異常。出血嚴重度與因子血中濃度相關性不佳。
- 非藥物治療:主要為單一捐贈者的新鮮冷凍血漿(fresh-frozen plasma, FFP),含所有凝血因子;缺點為容量過載、感染與抑制物風險。licensed for 血友病 B 的 PCCs 含維生素 K 依賴因子,亦可用於 RICD。
- 藥物治療:較輕的出血可單用抗纖溶胺基酸或合併因子補充,tranexamic acid 與 aminocaproic acid 可靜脈或口服給予,腎功能不全須調整劑量。
免疫性血小板低下症(ITP)#
流行病學與病理生理#
ITP 是後天血小板低下最常見原因之一,為免疫媒介的血小板破壞導致血小板壽命縮短。
- 兒童型:急性發作,常隨感染後出現,男女相當,4 至 6 週多自行緩解。
- 成人型:多為慢性(> 6 個月),女性為男性的 2 至 3 倍。
原發性 ITP 由自體免疫 IgG 抗體攻擊血小板表面醣蛋白;續發性 ITP 源於自體免疫疾病或感染(HIV、HCV、Helicobacter pylori)。
臨床表現為瘀點(petechiae)、紫斑(purpura)、瘀斑、黏膜出血、鼻出血、牙齦出血、血尿、經血過多,少見顱內出血。診斷依據血小板 < 100 × 10³/mm³、其餘血球與周邊抹片正常,並排除其他病因。
治療#
治療由是否出血及血小板數決定。在兒童多會自行緩解,部分情況無需治療。
- 成人主要目標:維持血小板 > 30 × 10³/mm³,低於此值出血風險增加。
- 第一線:prednisone(1 mg/kg/day)或高劑量 dexamethasone;皮質類固醇禁忌時用 anti-D immune globulin 或 IVIg。
- IVIg:阻礙 IgG 包覆的血小板被清除,約 80% 成人有反應但通常不持久,用於需快速提升血小板的情況。
- anti-Rh(D):僅用於未切脾的 Rh(D) 陽性病人,療效近似 IVIg 且較便宜。
- 第二線:脾切除(splenectomy)、rituximab(375 mg/m² 每週一次共四劑)、血小板生成素受體促效劑 eltrombopag 與 romiplostim(TPO mimetics)。
- 免疫抑制劑:azathioprine、cyclophosphamide 等保留給難治型。
eltrombopag 帶有肝毒性的黑框警示,需定期監測肝酵素;TPO mimetics 罕見但嚴重的風險包括骨髓變化、停藥後血小板低下惡化與血栓事件。
脾切除前 2 週應接種 Haemophilus influenzae b 型、肺炎鏈球菌與腦膜炎雙球菌疫苗,因主要併發症為細菌性敗血症。
血栓性微血管病變(TMA)#
概述#
TMA 包含多種病理機轉不同的疾病,典型表現為微血管病性溶血性貧血(microangiopathic hemolytic anemia)合併血小板低下,因小血管內皮缺陷致微血管血栓。原發性 TMA 包括血栓性血小板低下性紫斑(thrombotic thrombocytopenic purpura, TTP)、志賀毒素媒介溶血尿毒症候群、藥物誘發型、補體媒介型等。
實驗室特徵:ADAMTS13 活性下降(TTP)、血色素與血小板下降、周邊抹片見裂紅細胞(schistocytes)、haptoglobin 下降、LDH 與間接膽紅素上升、PT 與 aPTT 正常。
血栓性血小板低下性紫斑(TTP)#
TTP 因切割 vWF 的金屬蛋白酶 ADAMTS13 活性遺傳性缺乏或產生自體抑制物,使超大型 vWF 多聚體(ULvWF)累積,造成不當的血小板聚集與消耗。TTP 罕見造成腎臟異常,可與其他 TMA 區別。治療目標為預防末期器官損傷。
- 非藥物治療:後天型 TTP 採緊急血漿置換(plasma exchange, PEX)移除 ADAMTS13 抑制物並補充酵素,每日進行直到神經症狀緩解、LDH 與血小板維持正常數天。遺傳型則輸注含 ADAMTS13 的血漿。
- 藥物治療:glucocorticoids(如 methylprednisolone 或 prednisone)合併 PEX 提供免疫抑制;難治型可用 cyclosporine、rituximab。
補體媒介型 TMA#
源於補體替代路徑相關因子的基因突變,常由感染觸發,未受控的補體活化形成膜攻擊複合體造成內皮損傷、TMA 與腎衰竭。
- 支持治療:輸血、維持體液平衡、透析;肝腎移植可治癒但併發症顯著。
- 藥物治療:eculizumab 為人源化單株抗體,結合補體 C5 以阻止膜攻擊複合體形成,最好在發病 48 小時內開始。
eculizumab 會增加嚴重感染(含腦膜炎雙球菌)的風險,使用前應確認疫苗接種為最新狀態。
監測與結果評估#
- 血友病與 vWD:監測出血次數與類型、關節功能與影像評估、補充治療療效與抑制物的產生,並依標準接種疫苗。
- ITP:依臨床監測血小板數,成人維持 ≥ 30 × 10³/mm³,並監測出血徵象與藥物不良反應。
- TMA:當血小板維持 > 150 × 10³/mm³ 達 2 天可停止 PEX,glucocorticoids 可再續用 1 至 2 週;長期定期追蹤 CBC 與 LDH 以篩檢復發。