藥物動力學(pharmacokinetics)研究藥物的吸收(absorption)、分布(distribution)、代謝(metabolism,即生物轉化 biotransformation)與排除(elimination)——常合稱 ADME。理解並運用藥物動力學原理,可提高治療成功率並減少不良反應。要控制藥物的治療作用,必須知道有多少藥物到達作用部位、以及何時到達。
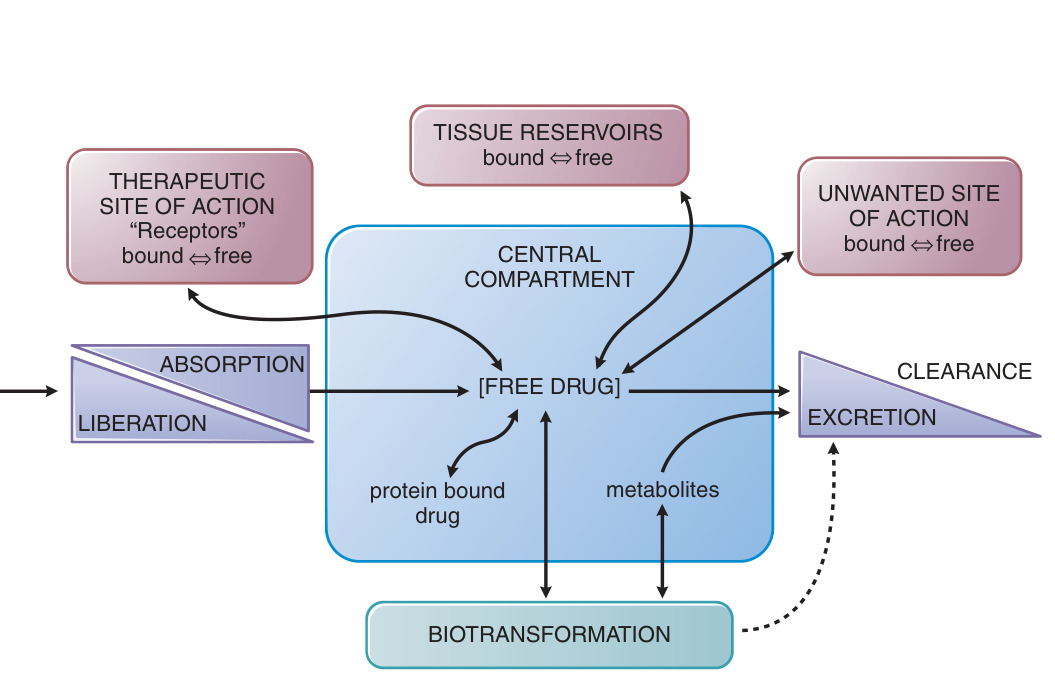
Figure 2–1:吸收、分布、結合、代謝與排泄的相互關係
藥物跨膜轉運的物理化學因素#
藥物的 ADME 全都涉及跨越細胞膜。決定藥物移動與在作用部位可用性的特性包括:分子大小與結構、解離程度、離子化與非離子化形式的脂溶性,以及與血漿和組織蛋白的結合。
細胞膜與被動擴散#
- 細胞膜由雙性脂質(amphipathic lipid)雙層構成,疏水尾朝內、親水頭朝外,對高極性分子相對不通透。膜蛋白可作為受體、離子通道或轉運體。膜並非均質流動,而是高度有序、區隔化的(如脂筏 lipid rafts、小窩 caveolae)。
- 多數藥物以**被動擴散(passive diffusion)**沿濃度梯度穿膜,速率正比於濃度梯度、脂水分配係數(partition coefficient)與膜表面積。
- 僅游離(未結合)藥物能跨膜;藥物與蛋白形成的複合物構成無活性的儲庫。
弱電解質與 pH 的影響#
許多藥物是弱酸或弱鹼,在溶液中同時以非離子化與離子化形式存在。
- 非離子化分子脂溶性較高、易擴散穿膜;離子化分子則較難穿透。
- 弱電解質的跨膜分布取決於其 pKa(半數藥物呈離子化時的 pH)與膜兩側的 pH 梯度,可用 Henderson-Hasselbalch 方程式描述。
- 穩態時,酸性藥物會累積在膜的較鹼側,鹼性藥物累積在較酸側(離子捕陷,ion trapping)。
此原理可用於藥物中毒處理:鹼化尿液(給予碳酸氫鈉)可促進弱酸(如阿斯匹靈 aspirin、尿酸)的排泄;酸化尿液則促進弱鹼的排泄。
載體媒介轉運#
- 主動轉運(active transport):需能量、可逆濃度梯度移動、具飽和性與選擇性,例如 Na+,K+-ATPase(毛地黃 digoxin 的治療標靶)。
- 次級主動轉運:利用既有梯度儲存的能量,如 Na+–Ca2+ 交換、鈉依賴性葡萄糖轉運體 SGLT1/SGLT2。
- 促進擴散(facilitated diffusion):無需能量、沿梯度移動,如 GLUT4。
- 重要的外排轉運體 P-醣蛋白(P-glycoprotein)(由
MDR1基因編碼)位於腸細胞,會將藥物排回腸腔以限制口服吸收,亦可賦予癌症化療抗藥性。
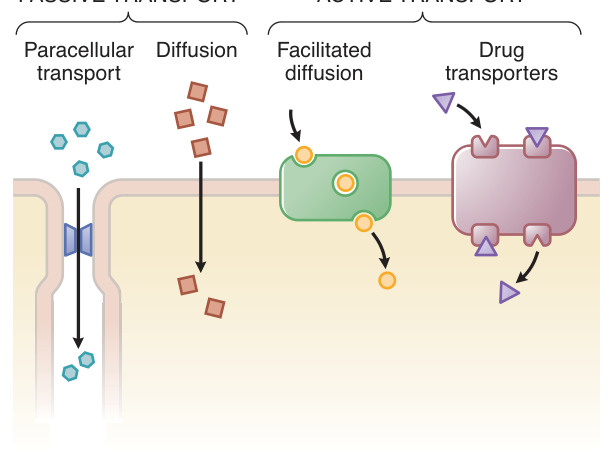
Figure 2–2:藥物跨細胞屏障的各種轉運方式
藥物吸收、生物利用度與給藥途徑#
吸收是藥物從給藥部位進入中央室的過程。臨床上更關注生物利用度(bioavailability,F)——藥物到達全身循環的比例。口服藥物經腸道吸收後,須先通過肝臟,部分會被腸道與肝臟代謝或經膽汁排泄,稱為首渡效應(first-pass effect)。
口服與腸道外給藥#
- 口服(oral):最常見、最安全、最方便、最經濟。缺點包括吸收受限、刺激胃腸黏膜、被消化酶或低胃酸破壞、需病人配合。
- 腸道外注射(parenteral):常較快速、完整且可預測,可精準給予有效劑量;在緊急狀況或病人無法口服時為必要。缺點是須無菌、可能疼痛、且注射後難以收回。
雖然依 pH 分配概念弱酸應在胃較易吸收,但小腸絨毛提供極大表面積(約 200 m²),故藥物在腸道的吸收速率通常大於胃。任何加速胃排空的因素都會增加吸收速率。
各種給藥途徑#
- 靜脈內(intravenous):繞過吸收限制,生物利用度完整且迅速、可精準控制。但高濃度可快速達成,可能引發不良反應,且注射後無退路。
- 皮下(subcutaneous):適合非刺激性藥物,吸收常恆定而緩慢;加入血管收縮劑(如 lidocaine 加 epinephrine)或植入固體可延長吸收。
- 肌肉內(intramuscular):水溶液吸收快,受局部血流影響;油性或儲庫製劑吸收緩慢持久。
- 舌下(sublingual):靜脈引流入上腔靜脈,繞過肝臟首渡代謝,如硝化甘油(nitroglycerin)。
- 經皮(transdermal):表皮為脂質屏障,吸收取決於面積與脂溶性;破損或發炎皮膚吸收增加。
- 直腸(rectal):約 50% 吸收的藥物可繞過肝臟。
- 肺部(pulmonary):表面積大、吸收迅速、避開肝臟首渡。
- 局部黏膜與眼睛:主要為局部作用,但可能發生全身吸收與毒性。
控釋製劑與生物等效性#
- 控釋(controlled-release)製劑設計為緩慢均勻吸收,可減少給藥頻率、維持夜間療效、降低峰谷波動,最適用於半衰期短(< 4 小時)的藥物。風險包括「劑量傾倒」(dose dumping)與被濫用(如 OxyContin)。
- 兩種藥品含相同活性成分、劑型、途徑且強度相同者為藥學等效;若兩者吸收速率與程度無顯著差異則為生物等效(bioequivalent)。晶型、粒徑等物理特性差異會影響吸收。
藥物分布#
吸收或全身給藥後,藥物分布至組織間液與細胞內液。心輸出量、區域血流、微血管通透性與組織體積決定分布速率與程度。肝、腎、腦等灌流良好的器官最先接受大部分藥物。
血漿蛋白與組織結合#
- **白蛋白(albumin)**主要結合酸性藥物;α1-酸性醣蛋白結合鹼性藥物。
- 結合通常可逆。低白蛋白血症(嚴重肝病、腎病症候群)會減少結合、增加游離分率。
- 只有游離藥物能跨膜並到達作用部位;血漿蛋白結合限制組織濃度與腎絲球過濾。
因疾病或藥物交互作用造成的蛋白結合改變,臨床上主要僅對少數靜脈給予、治療指數窄的高清除率藥物(如 lidocaine)有意義。
特殊儲庫與屏障#
- 脂肪:脂溶性藥物(如硫噴妥 thiopental)的儲庫,血流低而相對穩定。
- 骨骼:四環素、重金屬(如鉛、鐳)可累積於骨晶格。
- 再分布(redistribution):高脂溶性藥物(如硫噴妥)靜脈快速給藥後,先快速進入腦,再分布到肌肉等組織而終止作用,造成快速起效與快速終止。
- 血腦障壁(blood-brain barrier):腦微血管內皮有緊密接合,藥物須跨細胞轉運;脂溶性越高越易進入。P-醣蛋白與 OATP 等外排轉運體會限制藥物進入腦部。第二代抗組織胺(如 loratadine)腦濃度低故不嗜睡。
- 胎盤轉運:脂溶性、蛋白結合與解離程度決定通過程度;胎盤並非絕對屏障,胎兒會在某種程度上暴露於母親服用的所有藥物。
藥物的排除與代謝#
排除器官對極性化合物的排除效率高於高脂溶性物質。腎臟是排除藥物及其代謝物最重要的器官。
腎臟排泄#
涉及三個過程:
- 腎絲球過濾:僅游離藥物被過濾。
- 主動腎小管分泌:經 P-gp、MRP2(分泌結合代謝物)等轉運體。
- 被動腎小管再吸收:弱電解質的非離子化形式經非離子擴散再吸收,取決於尿液 pH。
膽汁與糞便排泄、腸肝循環#
- 肝細胞小管膜的轉運體(P-gp、BCRP、MRP2)將藥物與代謝物分泌入膽汁,釋入腸道。
- 腸肝循環(enterohepatic recycling):藥物與結合代謝物(如葡萄糖醛酸苷)經腸道菌酶水解後再吸收,可顯著延長藥物存留(如 ezetimibe 半衰期 > 20 小時)。
哺乳婦女服藥時,因乳汁較血漿酸,鹼性化合物可能在乳汁中略為濃縮,吸吮的嬰兒會在某種程度上暴露於藥物。
藥物代謝#
脂溶性有利穿膜卻不利排除,故須代謝為較親水的代謝物才能排除。
- 第一相反應(phase I):引入或暴露官能基(氧化、還原、水解),多由細胞色素 P450(CYP)催化,通常使藥理活性喪失。
- 第二相反應(phase II):與葡萄糖醛酸、硫酸鹽、麩胱甘肽等共軛,生成高極性、通常無活性的代謝物,便於排除。
- 前藥(prodrug):本身無活性,經代謝(常為酯或醯胺水解)轉為活性物,如 enalapril 轉為 enalaprilat。
- 代謝酶主要位於肝臟,其次為腸道、腎、肺。第一相酶多在內質網,第二相酶多在細胞質。
臨床藥物動力學#
臨床藥物動力學的基本前提是藥物的藥理作用與可量測濃度(如血漿)之間存在關係。四個最重要的參數為:生物利用度、分布體積、清除率與半衰期。
清除率(clearance, CL)#
清除率是設計長期給藥方案時最重要的概念,代表身體從全身循環清除藥物的效率。
- 假設完全生物利用,穩態時:給藥速率 = CL × Css(Css 為穩態濃度)。
- 清除率代表單位時間內被完全清除藥物的「血漿體積」,而非藥物量。各器官清除率可相加:CL = CL 腎 + CL 肝 + CL 其他。
- 多數藥物遵循一級動力學(first-order kinetics)——單位時間清除固定「分率」;若消除機制飽和,則趨近零級動力學——單位時間清除固定「量」(Michaelis-Menten)。
- 肝清除率:高萃取比藥物(如 propranolol、lidocaine)受肝血流限制;低萃取比藥物(如 warfarin)受蛋白結合與內在清除率影響。
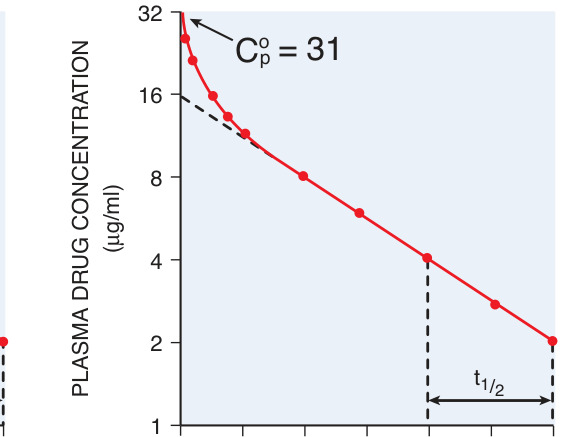
Figure 2–4:靜脈給藥後的血漿濃度-時間曲線
分布體積(volume of distribution, V)#
分布體積連結體內藥量與血漿濃度:V = 體內藥量 / C。它是「表觀」體積,不必對應實際生理空間。
- 高脂溶性或廣泛組織結合的藥物(如氯奎 chloroquine V 約 15,000 L、digoxin V 約 667 L)分布體積遠大於體液總量。
- 多室模型中常區分中央室(血液與灌流良好器官)與組織室。
半衰期(half-life, t½)與穩態#
- 半衰期是血漿濃度降低 50% 所需時間,t½ ≅ 0.693 × Vss / CL,同時受清除率與分布體積影響。
- 約經 4 個半衰期達到約 94% 的穩態;達到穩態的時間與劑量大小無關。
- 穩態濃度正比於劑量/給藥間隔(F/CL);峰谷波動正比於給藥間隔/半衰期。
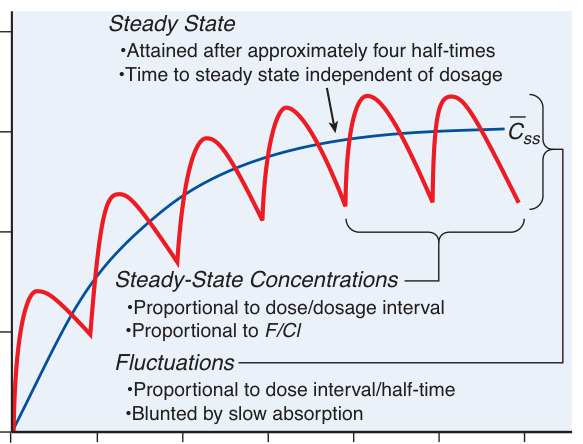
Figure 2–5:基本藥物動力學關係(穩態濃度與波動)
清除率下降(如腎衰竭)時 t½ 預期會延長,但此倒數關係僅在疾病未改變分布體積時成立。例如 diazepam 的 t½ 隨年齡增加,改變的其實是分布體積而非清除率。
非線性藥物動力學#
清除率、分布體積或 t½ 隨劑量改變,通常源於蛋白結合、肝代謝或腎轉運的飽和。
抗癲癇藥 phenytoin 的代謝在治療範圍內即飽和(Km 5–10 mg/L 接近治療範圍下緣)。劑量稍增即可能使濃度不成比例地、持續攀升而中毒;故治療指數窄且呈非線性代謝的藥物,治療藥物監測極為重要。
給藥方案的設計與最佳化#
藥物效果有特徵性的時間模式:強度與超過最低有效濃度(MEC, minimum effective concentration)的程度相關,持續時間反映濃度維持在 MEC 之上的時間。**治療窗(therapeutic window)**是兼具療效與可接受毒性的濃度範圍。
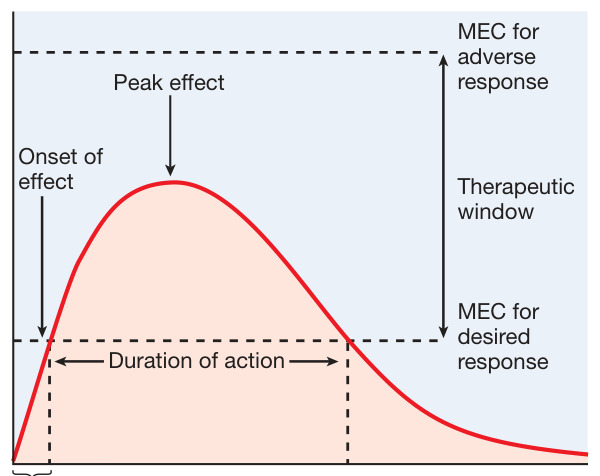
Figure 2–6:藥物效應的時間特性與治療窗
- 維持劑量:調整給藥速率使輸入等於損失,給藥速率 = 目標 Cp × CL / F。
- 負荷劑量(loading dose)= 目標 Cp × Vss / F,用於需快速達到治療濃度時(如心肌梗塞後使用 lidocaine)。缺點是敏感者可能突然暴露於毒性濃度,故常分次給予。
- 給藥間隔:間隔等於 t½ 時波動約 2 倍,通常可接受;低毒性藥物可採最大劑量策略、拉長間隔。
個體化劑量與治療藥物監測#
「一體適用」的標準方案忽略了顯著的個體間差異(F、CL、Vss 的標準差約 20%、50%、30%)。
- 對治療指數低的藥物(如心臟糖苷、抗心律不整藥、抗癲癇藥、免疫抑制劑、theophylline、warfarin),個體化劑量至關重要。
- 治療藥物監測(therapeutic drug monitoring):在穩態量測濃度以修正 CL/F 估計值。採樣應在前次給藥後、下次給藥前(濃度最低時)進行;給藥開始後須等約 4 個 t½ 達穩態才反映清除率。
- 早期吸收後的濃度反映吸收速率與中央室體積,對選擇長期維持劑量幾無意義。
「藥不吃就沒效。」順從性(compliance)是治療成敗的關鍵。未特別介入時,約僅 50% 病人合理遵循方案,約六分之一基本上不遵醫囑。漏服比過量常見,且每日須記得服藥的次數比藥物種類數更影響順從性;減少給藥次數可改善遵醫囑性。