藥物生物轉化概論#
大多數藥物在體內經過化學轉化(生物轉化,biotransformation),將其轉換為更易排出體外的產物。此過程通常將脂溶性、難以排泄的藥物轉化為水溶性代謝物,以利腎臟或膽汁排泄。
生物轉化不等於去活性化:
- 多數情況下:活性藥物 → 無活性代謝物(去活性)
- 某些情況:無活性前藥(prodrug)→ 活性藥物(活化,如可待因 codeine → 嗎啡 morphine)
- 某些情況:藥物 → 毒性代謝物(如乙醯胺酚 acetaminophen → NAPQI)
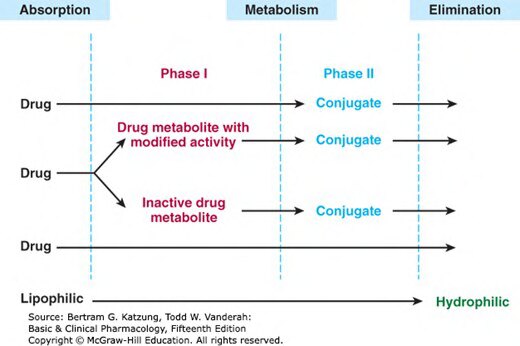
Figure 4-1:第一相與第二相反應及直接排除途徑示意圖,第二相反應亦可先於第一相發生
第一相反應(Phase I Reactions)#
第一相反應通常引入或暴露官能基(-OH、-NH₂、-SH、-COOH),使藥物極性增加,或為第二相反應提供基質。
氧化反應(Oxidation)#
細胞色素 P450(CYP)酶系統 是最重要的氧化代謝酶,位於肝臟(主要)、腸壁、肺臟等組織的內質網(endoplasmic reticulum)。
P450 催化反應的基本步驟:
- 藥物(RH)與 CYP 的 Fe³⁺ 結合
- 從 NADPH 接受電子,Fe³⁺ 還原為 Fe²⁺
- Fe²⁺ 與 O₂ 結合,形成活性氧複合物
- 一個氧原子插入底物(形成 R-OH),另一個氧原子形成水
- Fe³⁺ 再生,繼續催化下一個循環
反應整體:RH + O₂ + NADPH + H⁺ → ROH + H₂O + NADP⁺
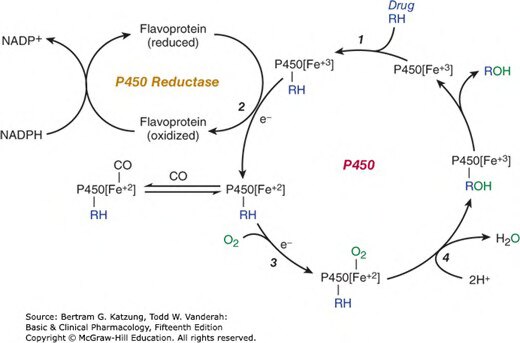
Figure 4-3:細胞色素 P450 催化藥物氧化的循環機制,e⁻ 為電子,RH 為原藥,ROH 為氧化代謝物
非 P450 的氧化酶還包括:黃素單加氧酶(FMO)、單胺氧化酶(MAO)、醇脫氫酶(ADH)、醛脫氫酶(ALDH)。
還原反應(Reduction)與水解反應(Hydrolysis)#
- 還原:如氯黴素(chloramphenicol)的硝基還原
- 水解:酯酶(esterases)和醯胺酶(amidases)水解酯鍵或醯胺鍵,如阿斯匹靈(aspirin)→ 水楊酸、普魯卡因(procaine)→ 對氨基苯甲酸
主要 CYP 同功酶#
人類肝臟中最重要的藥物代謝 CYP 同功酶:
| 同功酶 | 主要底物(代表藥物) | 主要誘導劑 | 主要抑制劑 |
|---|---|---|---|
| CYP1A2 | 咖啡因、茶鹼、氯氮平 | 吸菸、烤肉(多環芳烴) | 氟伏沙明 |
| CYP2C9 | 華法林(S 型)、NSAIDs、苯妥英 | 利福平 | 氟康唑、胺碘酮 |
| CYP2C19 | 奧美拉唑、氯吡格雷(前藥)、某些 TCAs | 利福平 | 奧美拉唑、氟西汀 |
| CYP2D6 | 可待因、三環抗憂鬱劑、β-blockers、抗精神病藥 | (幾乎不被誘導) | 奎尼丁、帕羅西汀 |
| CYP2E1 | 乙醇、乙醯胺酚(高劑量)、某些麻醉劑 | 乙醇、異煙肼 | 二硫龍 |
| CYP3A4/5 | 大多數藥物(> 50%):環孢素、他克莫司、他汀類、苯二氮平、大環內酯類 | 利福平、卡馬西平、聖約翰草 | 伊曲康唑、利托那韋、葡萄柚汁 |
CYP3A4 是最重要的藥物代謝酶,代謝超過半數的上市藥物,同時也是最多藥物交互作用的根源。
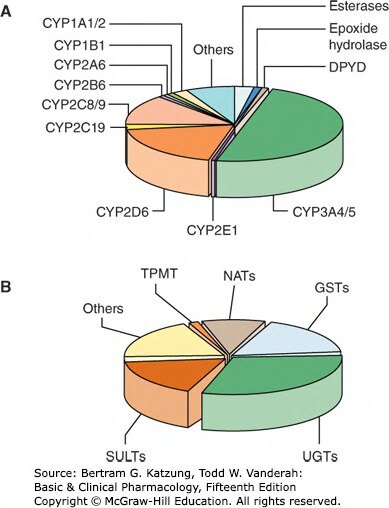
Figure 4-4:參與藥物代謝的第一相(A,CYP 家族為主)與第二相(B,UGT/SULT/GST 等)酶系各占比例圓餅圖
第二相反應(Phase II Reactions)#
第二相反應是結合反應(conjugation),將含官能基的藥物或其第一相代謝物與內源性極性基團共價連接,使產物高度水溶,幾乎都失去活性並易於排泄。
| 反應類型 | 供體 | 酶 | 代表底物 |
|---|---|---|---|
| 葡萄糖醛酸化(glucuronidation) | UDP-葡萄糖醛酸(UDPGA) | UGT(尿苷二磷酸葡萄糖醛酸轉移酶) | 嗎啡、乙醯胺酚、膽紅素 |
| 乙醯化(acetylation) | 乙醯 CoA | NAT(N-乙醯基轉移酶) | 異煙肼、磺胺藥、普魯卡因胺 |
| 硫酸化(sulfation) | PAPS | SULT(磺基轉移酶) | 甲基多巴、類固醇 |
| 甲基化(methylation) | S-腺苷甲硫氨酸(SAM) | COMT、TPMT | 多巴胺、6-硫嘌呤 |
| 穀胱甘肽結合(glutathione conjugation) | 穀胱甘肽(GSH) | GST(穀胱甘肽-S-轉移酶) | NAPQI(乙醯胺酚毒性代謝物)、活性中間體 |
酶的誘導(Enzyme Induction)#
某些藥物、食物或環境因素可增加 CYP 酶的表現量,加速底物藥物的代謝,降低其血漿濃度和效應。
誘導機制#
- AhR(芳烴受體)路徑:多環芳烴(吸菸)、TCDD 活化 AhR → 誘導 CYP1A1/1A2
- PXR(孕烷 X 受體)路徑:利福平(rifampin)、卡馬西平(carbamazepine)、苯妥英、聖約翰草(St. John’s Wort)→ 誘導 CYP3A4、CYP2C9、P-gp
- CAR(組成型雄烷受體)路徑:苯巴比妥(phenobarbital)→ 誘導 CYP2B6、CYP3A4
臨床意義#
酶誘導導致的藥物交互作用:
- 利福平 + 口服避孕藥:CYP3A4 誘導使避孕藥濃度降低 → 避孕失效
- 利福平 + 華法林:CYP2C9 誘導 → 華法林抗凝效果減弱,需大幅增加劑量
- 聖約翰草 + 環孢素(cyclosporine):CYP3A4 誘導 → 移植排斥風險升高
- 停用誘導劑後,酶活性恢復需 1–4 週
酶的抑制(Enzyme Inhibition)#
CYP 抑制 使底物藥物代謝減緩,血漿濃度升高,可能導致毒性。
類型#
- 競爭性抑制(competitive inhibition):抑制劑與底物競爭同一結合位點;效果立即,停藥後消失快
- 機制基礎性抑制(mechanism-based inhibition, MBI)/ 不可逆性抑制:抑制劑先被 CYP 代謝成活性中間體,再與酶共價結合,使酶永久失活。酶活性的恢復需等待新酶合成(數天至一週)
機制基礎性 CYP 抑制劑(如克拉黴素 clarithromycin、紅黴素 erythromycin、地爾硫卓 diltiazem、葡萄柚汁的呋喃香豆素 furanocoumarins)的相互作用具有時間延遲性——即使停藥,效果仍可持續數天,因此風險期比競爭性抑制劑更長。
重要抑制劑#
- 伊曲康唑(itraconazole)、酮康唑(ketoconazole):強效 CYP3A4 抑制劑
- 利托那韋(ritonavir):極強效 CYP3A4 抑制劑,在 HIV 雞尾酒療法中用作藥動學「增強劑」(booster)以提高其他蛋白酶抑制劑濃度
- 奎尼丁(quinidine):強效 CYP2D6 抑制劑
- 葡萄柚汁(grapefruit juice):選擇性抑制腸道 CYP3A4(不影響肝臟),增加某些藥物的口服生物可用性(如辛伐他汀 simvastatin、非洛地平 felodipine)
代謝為毒性產物:乙醯胺酚肝毒性#
乙醯胺酚(acetaminophen,又稱 paracetamol) 的肝毒性是藥物代謝生成毒性產物的經典案例:
正常劑量下:
- ~90% 經葡萄糖醛酸化(UGT)及硫酸化,形成無毒性的水溶性結合物由腎臟排出
- ~5–10% 經 CYP2E1(少量由 CYP3A4)氧化,生成高度活性的毒性中間體 N-乙醯-對苯醌亞胺(NAPQI)
- NAPQI 立即被穀胱甘肽(GSH) 解毒,形成無毒結合物
過量時:
- 硫酸化途徑飽和,更多藥物進入 CYP 氧化途徑
- 大量 NAPQI 生成,消耗 GSH 儲備
- GSH 耗盡後,NAPQI 與肝細胞蛋白質共價結合 → 肝細胞壞死
乙醯胺酚肝毒性的危險因素:
- 過量使用(成人單次 > 7.5 g 或慢性 > 4 g/d)
- 長期大量飲酒(誘導 CYP2E1,並耗盡 GSH)
- 禁食或營養不良(GSH 合成原料不足)
- 解毒劑:N-乙醯半胱胺酸(N-acetylcysteine, NAC),可補充 GSH 前驅物,在過量後 8 小時內給予效果最佳。
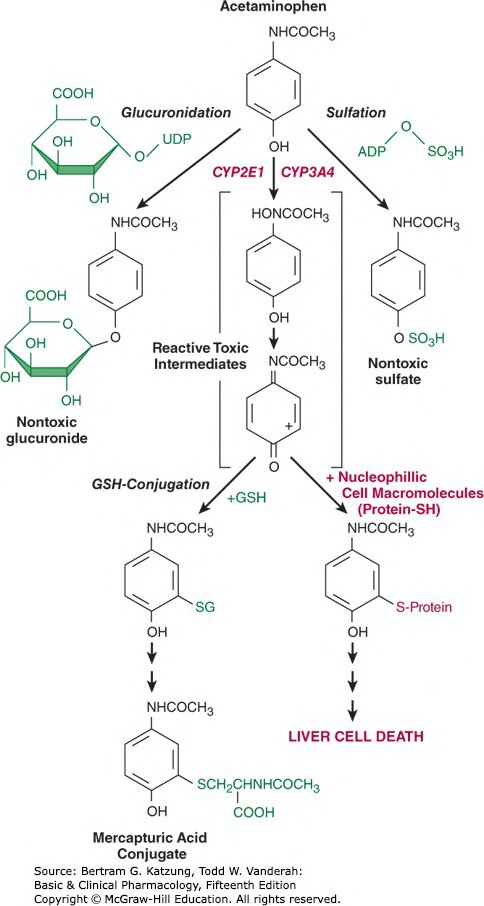
Figure 4-5:乙醯胺酚代謝途徑及其肝毒性機制,包括葡萄糖醛酸化、硫酸化、CYP2E1/3A4 氧化生成 NAPQI,以及 GSH 解毒與蛋白質共價結合導致肝細胞死亡
遺傳多型性對藥物代謝的影響#
CYP2D6 多型性#
CYP2D6 代謝約 25% 的常用藥物,基因多型性影響顯著:
- 慢代謝者(PM, poor metabolizers):功能缺失等位基因,約 5–10% 白人
- 可待因(codeine)→ 嗎啡的轉化受阻,鎮痛效果差
- 三環抗憂鬱劑(TCAs)代謝減慢,濃度升高,毒性風險增加
- 超快代謝者(UM, ultrarapid metabolizers):基因拷貝數增加,約 1–5% 白人,20%+ 北非人
- 可待因 → 嗎啡轉化過多 → 嗎啡累積 → 呼吸抑制,嬰兒哺乳死亡案例
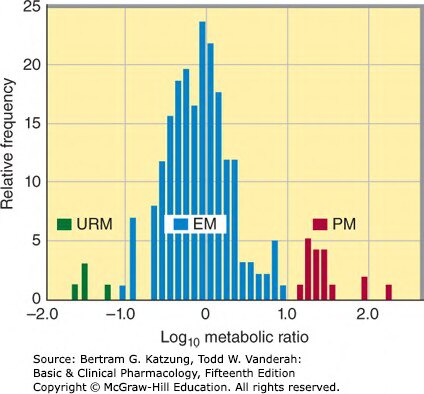
Figure 4-6:CYP2D6 代謝多型性分布直方圖,顯示人群中超快代謝者(URM)、正常代謝者(EM)與慢代謝者(PM)的 log₁₀ 代謝比率分布
CYP2C19 多型性#
- 代謝物質:質子幫浦抑制劑(PPIs)、氯吡格雷(前藥)
- PM 約 2–5% 白人,15–20% 亞洲人
- 氯吡格雷(clopidogrel)為前藥,需 CYP2C19 活化:PM 患者活性代謝物不足,抗血小板效果差,心血管事件風險升高
CYP2C9 多型性#
- 代謝物質:S-華法林(warfarin)、NSAIDs、苯妥英
- CYP2C9*2 和 *3 等位基因使酶活性降低 30–90%
- 攜帶 *2/*3 等位基因者需要較低的華法林劑量以避免出血
第二相反應的多型性#
NAT2(N-乙醯基轉移酶 2)乙醯化多型性:
- 慢乙醯化者(slow acetylators):異煙肼(isoniazid)代謝慢 → 血漿濃度高 → 周圍神經病變(peripheral neuropathy)風險高
- 快乙醯化者:藥物清除快 → 抗結核療效可能不足
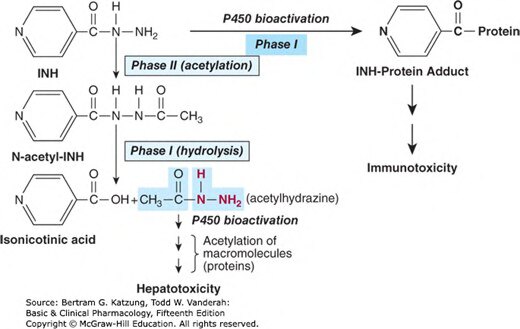
Figure 4-2:異煙肼(INH)的代謝途徑,包括第二相乙醯化(N-acetyl-INH)與第一相 P450 生物活化,分別導致免疫毒性與肝毒性
TPMT(硫嘌呤甲基轉移酶)多型性:
- 活性低者(~1 in 300 人):6-巰嘌呤(6-mercaptopurine)、硫唑嘌呤(azathioprine)代謝受阻 → 骨髓毒性風險極高
UGT1A1 多型性:
- UGT1A1*28 等位基因:伊立替康(irinotecan)的活性代謝物 SN-38 葡萄糖醛酸化減少 → 嚴重腹瀉和中性球低下風險升高
腸道微生物的作用#
腸道菌群(gut microbiota)含有多種代謝酶,可影響部分藥物的代謝:
- 地高辛(digoxin)在某些個體中被腸道 Eggerthella lenta 菌大量代謝,使口服生物可用性降低
- 硫酸結合物在腸道中被菌群水解(腸肝循環),延長某些藥物的作用時間
- 腸道菌群差異可能解釋某些藥物反應的個體差異
飲食與環境因素#
- 吸菸:誘導 CYP1A2,降低氯氮平(clozapine)、茶鹼(theophylline)等血漿濃度
- 葡萄柚汁:抑制腸道 CYP3A4,增加辛伐他汀、非洛地平等藥物的生物可用性(可增加數倍)
- 十字花科蔬菜(如綠花椰菜):含吲哚化合物,誘導 CYP1A2
疾病對藥物代謝的影響#
- 肝臟疾病:肝硬化(cirrhosis)使肝臟代謝酶量和肝血流量同時下降,高萃取率藥物(如利多卡因)影響尤其顯著
- 心臟衰竭:心輸出量下降 → 肝血流量減少 → 高萃取率藥物代謝減少
- 腎衰竭:除影響腎排泄外,也可影響某些代謝酶活性和蛋白結合率
大分子生物製劑的代謝#
蛋白質、多肽藥物和單株抗體(mAbs) 不經由 CYP 代謝,而是透過:
- 蛋白酶水解(降解為胺基酸)
- FcRn(新生兒 Fc 受體) 循環:IgG 型 mAbs 與 FcRn 結合,被從溶酶體中拯救,延長半衰期(長達 2–3 週)
- 靶點介導的藥物分佈與消除(TMDD):高親和力藥物在靶點飽和前,清除速率隨藥物濃度大幅變化
生物製劑不發生 CYP 相關的藥物交互作用,但嚴重炎症(如 IL-6 升高)可透過下調 CYP3A4 影響小分子藥物的代謝——此現象在使用 tocilizumab 等 IL-6 阻斷劑後可逆轉,需調整同時使用的小分子藥物劑量。
小結#
藥物生物轉化分為兩相:第一相(氧化/還原/水解)引入官能基,第二相(結合反應)增加水溶性。CYP P450 酶系是第一相代謝的核心,其中 CYP3A4、CYP2D6、CYP2C9、CYP2C19 代謝了大多數上市藥物。酶的誘導(如利福平誘導 CYP3A4)和抑制(如葡萄柚汁抑制 CYP3A4)是重要藥物交互作用的根源。遺傳多型性(CYP2D6、CYP2C19、CYP2C9、NAT2、TPMT)導致個體間代謝能力的顯著差異,影響藥物療效與毒性。