藥動學概論#
藥動學(pharmacokinetics)研究身體如何處置藥物,即藥物在體內的吸收(absorption)、分佈(distribution)、代謝(metabolism)與排泄(excretion),合稱 ADME。
藥動學的核心目標是理解「藥物濃度如何隨時間變化」,以便合理設計給藥方案,使藥物濃度維持在**治療窗口(therapeutic window)**內:高於最低有效濃度、低於最低毒性濃度。
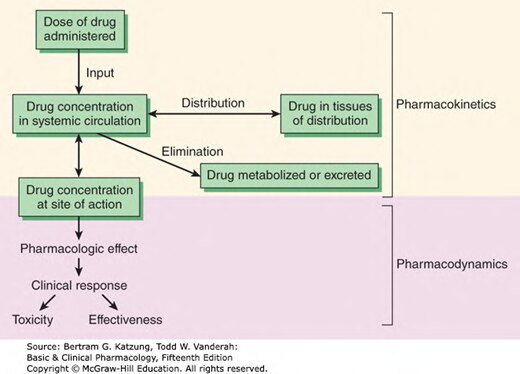
Figure 3-1:藥動學與藥效學的關係——從給藥劑量到藥理效應與臨床反應的流程圖
基本藥動學參數#
表觀分佈容積(Volume of Distribution, Vd)#
Vd 是藥物在體內「假設均勻分佈」所需的理論容積:
Vd = 藥物總量(D) / 血漿濃度(C)- Vd 小(接近血漿容積 ~3 L):藥物主要留在血管內,如肝素(heparin)
- Vd 大(數百至數千升):藥物大量分佈至組織中,如氯喹(chloroquine,~15,000 L)
Vd 是表觀值,不代表真實的生理容積。Vd 大的藥物不易被血液透析清除,因為藥物主要在組織中。
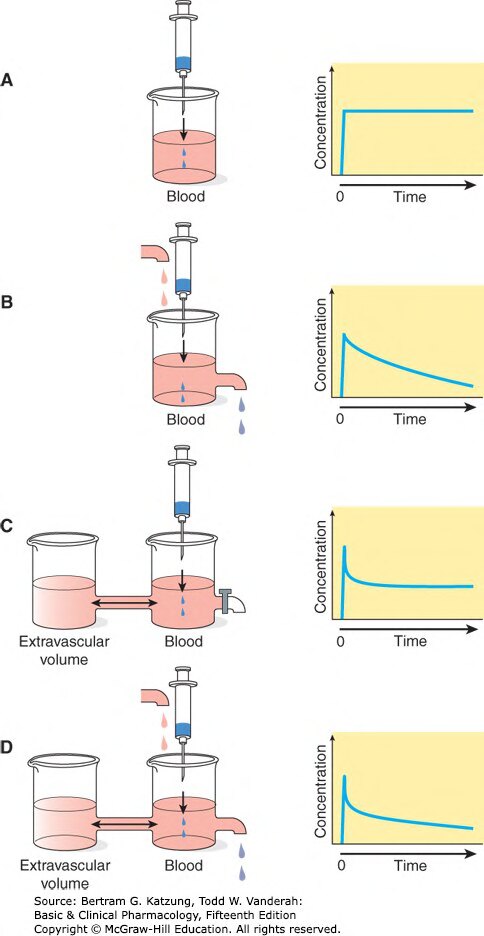
Figure 3-2:以隔室模型示意分佈容積(Vd)的概念——從單一血液隔室到含外血管隔室的四種情境
清除率(Clearance, CL)#
CL 是單位時間內被「清除」(代謝或排泄)的藥物所相當的血漿容積:
CL = 消除速率 / 血漿濃度- 清除率的單位:L/h 或 mL/min
- 主要清除器官:肝臟(代謝)、腎臟(排泄)
- 全身清除率 = 肝臟清除率 + 腎臟清除率 + 其他
半衰期(Half-Life, t½)#
t½ 是血漿藥物濃度下降 50% 所需的時間:
t½ = 0.693 × Vd / CL- 半衰期決定藥物的給藥頻率(通常每 1–2 個半衰期給藥一次)
- 達到穩態(steady state)需要約 4–5 個半衰期
- 停藥後約 4–5 個半衰期藥物基本清除
半衰期由 Vd 和 CL 共同決定。腎功能不全時 CL 下降,t½ 延長,藥物可能蓄積至毒性濃度,需調整劑量或給藥間隔。
穩態(Steady State)#
以固定劑量、固定間隔重複給藥時,輸入速率與消除速率最終相等,血漿濃度在某個範圍內波動(峰值 Cmax 和谷值 Cmin 之間),稱為穩態:
- 達到穩態所需時間 = 4–5 個半衰期(與劑量無關)
- 穩態平均濃度(Css_avg)= 給藥速率 / CL
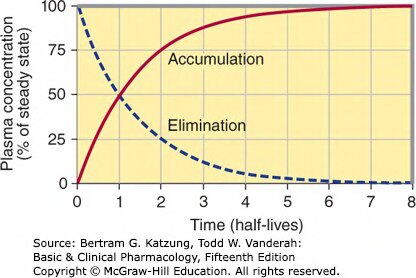
Figure 3-3:反覆給藥時藥物蓄積(Accumulation)與停藥後消除(Elimination)曲線——以半衰期為橫軸,佔穩態百分比為縱軸
生物可用性(Bioavailability, F)#
F 是藥物到達體循環的分率(靜脈注射 F = 1,即 100%):
F = AUC(口服)/ AUC(靜脈注射)× 100%影響口服藥物生物可用性的因素:
- 溶解度與吸收(腸道 pH、轉運蛋白)
- 首過效應(first-pass effect):口服藥物經腸壁和肝臟代謝,導致進入體循環前大量被消除
- 腸道 P-糖蛋白(P-gp)外排
高首過效應的藥物(如硝酸甘油 nitroglycerin、普萘洛爾 propranolol)口服生物可用性低,硝酸甘油須舌下給藥(sublingual)以繞過首過效應。
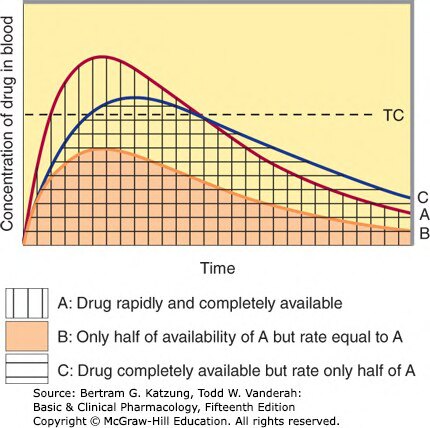
Figure 3-4:不同生物可用性情境(A/B/C)的血漿濃度時間曲線比較——展示藥量完整可用、藥量減半及吸收速率減半對 AUC 與 Cmax 的影響
給藥途徑#
| 途徑 | 優點 | 缺點 |
|---|---|---|
| 口服(PO) | 方便、安全 | 首過效應、吸收受腸道影響 |
| 靜脈注射(IV) | F=100%、起效快 | 侵入性、感染風險 |
| 肌肉注射(IM) | 吸收可靠 | 疼痛、吸收速率可變 |
| 皮下注射(SC) | 適用蛋白質藥物 | 吸收速率慢 |
| 舌下(SL) | 繞過首過效應 | 只適合脂溶性、小分子藥物 |
| 透皮貼片(TD) | 持續給藥、無首過效應 | 吸收速度慢、受皮膚影響 |
| 吸入(Inhalation) | 局部作用、起效快 | 技術要求高 |
藥物在體內的時間過程#
單次靜脈注射後#
單次 IV 注射後,血漿濃度以指數函數下降:
C(t) = C₀ × e^(-kel × t)其中消除速率常數 kel = CL / Vd = 0.693 / t½。
口服給藥後#
血漿濃度先上升(吸收大於消除)後下降(消除大於吸收),形成峰值(Cmax):
- Tmax:達到 Cmax 的時間
- AUC(曲線下面積)反映藥物總暴露量,與 F 和劑量成正比
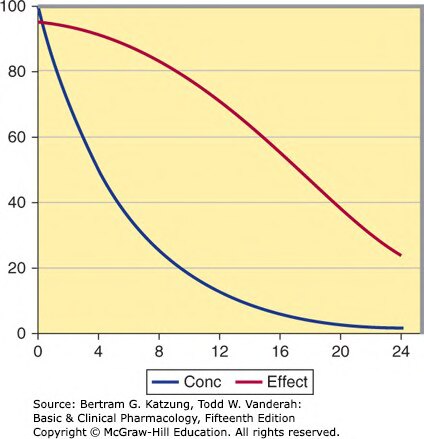
Figure 3-5:血漿濃度(Conc)與藥理效應(Effect)隨時間的對比曲線——效應消退明顯滯後於濃度下降
目標濃度策略(Target Concentration Strategy)#
合理用藥的基礎是設定目標血漿濃度,再據此計算給藥方案。
維持劑量(Maintenance Dose)#
在穩態時,給藥速率須等於消除速率:
給藥速率(Dosing Rate)= 目標 Css × CL / F- CL 因病人而異(腎功能、肝功能、遺傳多型性)
- 腎功能不全時 CL 降低,需減少劑量或延長給藥間隔
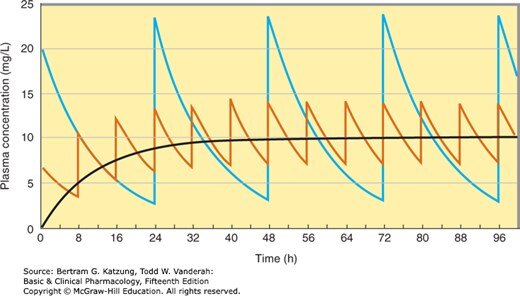
Figure 3-6:反覆口服給藥的血漿濃度時間曲線——比較不同給藥間隔(8 h vs 24 h)下的濃度波動與穩態平均濃度
負荷劑量(Loading Dose)#
對於半衰期長的藥物,若急需快速達到治療濃度,可先給予較大的負荷劑量(loading dose):
Loading Dose = 目標 Css × Vd / F地高辛(digoxin)半衰期約 36 小時,若只從維持劑量開始,需一週多才達穩態。臨床急症時可先給負荷劑量(但需注意毒性)。
負荷劑量的適用情境:
- 藥物 t½ 長(> 12 小時)
- 需要快速起效(如心房顫動(atrial fibrillation)的速率控制)
- Vd 大的藥物(負荷劑量也較大)
劑量個人化(Dose Individualization)#
影響藥物清除率的因素#
腎功能:
- 腎臟排泄為主的藥物(如胺糖苷類 aminoglycosides、萬古黴素 vancomycin、地高辛)需根據腎小球過濾率(GFR)調整
- 常用 Cockcroft-Gault 公式或 CKD-EPI 估算肌酐清除率(creatinine clearance, CrCl)
肝功能:
- 高肝臟萃取率(extraction ratio > 0.7)的藥物:清除率受肝血流量影響(如利多卡因 lidocaine、普萘洛爾)
- 低肝臟萃取率(< 0.3)的藥物:清除率受肝臟固有酶活性及血漿蛋白結合率影響(如華法林 warfarin、苯妥英 phenytoin)
年齡:
- 新生兒:肝臟代謝酶(尤其 UGT)及腎小管分泌功能未成熟,多數藥物 t½ 延長
- 老年人:CL 下降(肌肉量減少使肌酐低估 GFR)、Vd 改變(體脂增加)、蛋白結合降低
遺傳因素:
- CYP2D6、CYP2C19 等代謝酶的基因多型性可使個體間 CL 差異達 10 倍以上
治療藥物監測(Therapeutic Drug Monitoring, TDM)#
TDM 適用於:
- 治療窗口窄的藥物(如地高辛、鋰鹽、抗癲癇藥、胺糖苷類抗生素)
- 藥物效應難以直接量測(如預防性抗凝、預防癲癇發作)
- 懷疑藥物交互作用或毒性
採血時機很重要:
- 谷值(trough):下次給藥前採血,反映最低暴露量,最常用
- 峰值(peak):給藥後特定時間採血,用於評估毒性(如胺糖苷類的腎毒性監測)
- 採血前須確認病人已達穩態(至少 4–5 個 t½ 後)
藥物濃度的解讀#
血漿濃度測定值需結合臨床情境解讀:
- 濃度「正常」但無效 → 懷疑吸收問題、游離(未結合)分率異常、耐藥性
- 濃度「正常」但有毒性 → 懷疑蛋白結合率降低(低白蛋白血症時,游離藥物比例升高)
- 濃度「低」→ 確認用藥依從性(compliance)、藥物交互作用(誘導)、吸收障礙
苯妥英(phenytoin)在低白蛋白血症(如腎衰竭、肝硬化)病患中,測得的「總濃度」偏低,但游離型(具藥理活性)濃度可能正常甚至偏高。應使用 Sheiner-Tozer 公式校正,或直接測定游離苯妥英濃度。
非線性藥動學(Nonlinear Pharmacokinetics)#
大多數藥物遵循線性(first-order)藥動學:消除速率與濃度成正比,t½ 固定。
少數藥物在治療濃度時,代謝酶或轉運蛋白已接近飽和,呈現非線性(zero-order 或 Michaelis-Menten)藥動學:
- 代表藥物:苯妥英(phenytoin)、乙醇(alcohol)、阿斯匹靈(高劑量)
- 小幅度增加劑量可導致血漿濃度大幅上升 → 毒性風險高
- TDM 在此類藥物尤為重要
苯妥英的治療濃度(10–20 mg/L)與其 Km 接近,一旦劑量稍過,代謝飽和後濃度急遽上升,易發生毒性(眼球震顫、步態不穩、意識混亂)。劑量調整應謹慎,每次小幅調整後須再次監測。
小結#
藥動學的核心參數——分佈容積(Vd)、清除率(CL)、半衰期(t½)、生物可用性(F)——共同決定藥物在體內的時間過程。合理給藥方案的設計基於目標濃度策略:維持劑量由清除率決定,負荷劑量由分佈容積決定;半衰期決定達到穩態所需的時間與給藥頻率。個體差異(腎肝功能、年齡、遺傳)使劑量個人化成為必要,治療藥物監測是窄治療窗口藥物安全使用的重要工具。