藥效學概論#
藥效學(pharmacodynamics)研究藥物對身體的效應,核心問題是:藥物如何透過與特定分子靶點結合,產生可量化的生物效應?
受體的巨分子本質#
什麼是受體?#
受體(receptor) 是藥物或內源性配體(endogenous ligand)結合並啟動生物效應的特定巨分子,通常為蛋白質。受體的基本性質:
- 特異性(specificity):受體對特定配體有高度選擇性
- 飽和性(saturability):受體數量有限,高濃度下效應達到上限
- 可逆性(reversibility):多數藥物-受體結合是可逆的(共價結合者除外)
受體的種類#
細胞內受體或細胞膜上的各類蛋白質均可作為藥物靶點,包括:
- 酶(enzymes):如 HMG-CoA 還原酶(reductase)是史他汀(statins)的靶點
- 離子通道(ion channels):如局部麻醉藥阻斷電壓依賴性鈉通道
- 轉運蛋白(transporters):如選擇性血清素再回收抑制劑(SSRIs)阻斷 SERT
- 訊號受體(signaling receptors):包含 G 蛋白偶合受體、酪氨酸激酶受體等
並非所有藥物都透過特異性受體作用。例如:制酸劑(antacids)透過化學中和作用;滲透性利尿劑透過物理性效果;揮發性麻醉劑可能透過非特異性膜效應發揮作用。
濃度-效應關係#
量效曲線(Concentration-Effect Curves)#
藥物效應通常與受體占據率(receptor occupancy)相關。以藥物濃度(對數值)對效應作圖,得到 S 型(sigmoid)曲線:
- EC₅₀:產生最大效應 50% 所需的藥物濃度,代表藥物效價(potency)
- E_max:藥物所能產生的最大效應,代表藥物效能(efficacy)
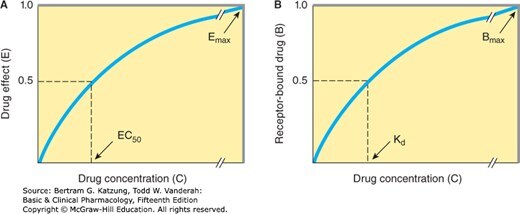
Figure 2-1:藥物效應(E)與受體結合量(B)對濃度的雙曲線,標示 EC₅₀、E_max 及解離常數 Kd
- 效價高代表需要的劑量少(EC₅₀ 小)
- 效能高代表可以達到的最大效應大(E_max 高) 臨床上效能通常比效價更重要。
解離常數(Kd)與受體親和力#
藥物(D)與受體(R)的結合反應:
D + R ⇌ DR解離常數 Kd = [D][R] / [DR],Kd 越小表示親和力越高。
備用受體(Spare Receptors)#
某些組織中,達到最大效應(E_max)所需的受體占據率遠低於 100%,這些「多餘」的受體稱為備用受體(spare receptors)。
- 備用受體的存在使得 EC₅₀(功能上)可以遠低於 Kd(結合上)
- 備用受體提高了組織對配體的敏感性,是安全機制之一
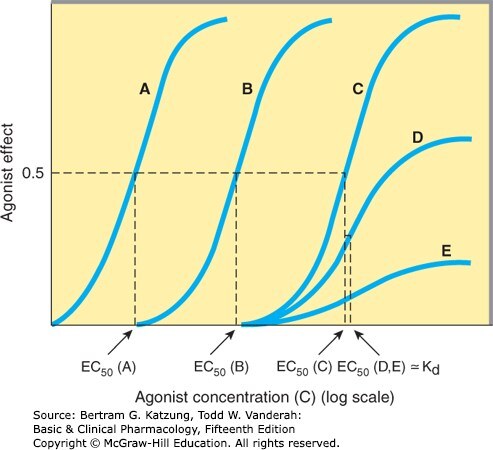
Figure 2-2:多條對數 S 型量效曲線比較不同激動劑的效價與備用受體效應(EC₅₀ vs Kd 的差距反映備用受體存在)
拮抗劑與部分激動劑#
競爭性拮抗劑(Competitive Antagonists)#
競爭性拮抗劑與激動劑競爭同一結合位點:
- 效應可被增加激動劑濃度所克服
- 使量效曲線右移,但 E_max 不變
- 以拮抗劑常數(Ki 或 pA₂) 衡量其效價
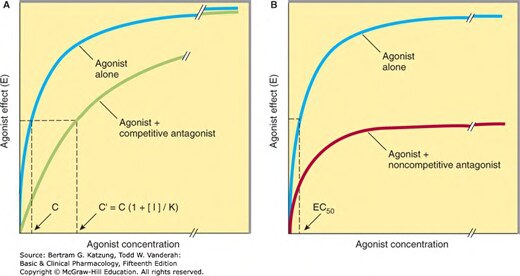
Figure 2-3:競爭性拮抗劑使量效曲線右移(E_max 不變)vs 非競爭性拮抗劑使 E_max 下降的比較
不可逆性拮抗劑(Irreversible Antagonists)#
與受體形成共價或極高親和力的非共價結合:
- 拮抗效應不能被激動劑濃度增加所逆轉
- 使量效曲線右移,且 E_max 下降
- 代表藥物:苯氧苯扎明(phenoxybenzamine,不可逆性 α 受體拮抗劑)
在有備用受體的系統中,不可逆性拮抗劑在低濃度時(僅占據少量受體)可能只造成右移而不降低 E_max;當受體被大量占據超過備用受體的比例時,E_max 才開始下降。
部分激動劑(Partial Agonists)#
部分激動劑結合受體後,其 E_max 低於完全激動劑(full agonist):
- 在無完全激動劑的情況下,表現為弱激動作用
- 在有完全激動劑的情況下,因競爭同一受體而表現為拮抗作用
- 代表藥物:丁丙諾啡(buprenorphine,部分 μ 阿片受體激動劑)
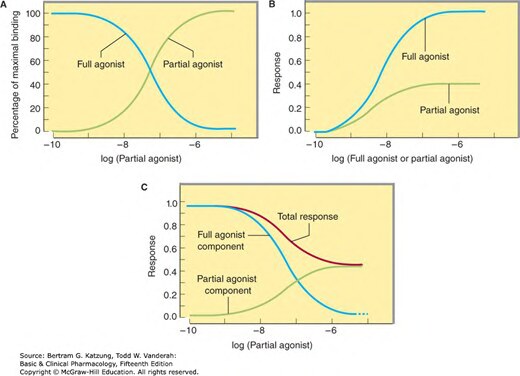
Figure 2-4:部分激動劑與完全激動劑的結合曲線(A)、個別效應曲線(B),及混合存在時的總效應(C)
跨膜訊號傳遞機制#
細胞跨膜訊號傳遞有五大主要類型:
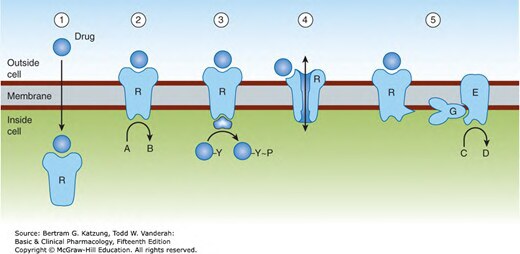
Figure 2-5:五大跨膜訊號傳遞機制示意圖——細胞內受體(1)、酶聯受體(2/3)、離子通道(4)、G 蛋白偶合受體(5)
機制一:細胞內受體(Intracellular Receptors)#
脂溶性配體(如類固醇激素、甲狀腺素)可穿越細胞膜,與細胞質或細胞核內的受體結合:
- 受體-配體複合物作為轉錄因子(transcription factors) 調控基因表現
- 效應起效慢(小時至天),持續時間長
- 例如:糖皮質素(glucocorticoids)與 GR(糖皮質素受體)結合,調控抗炎基因
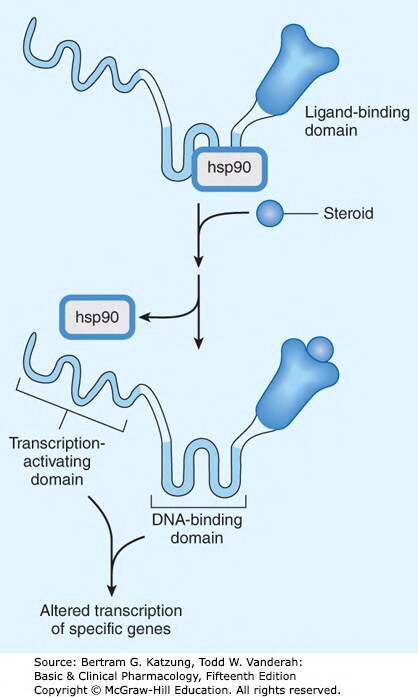
Figure 2-6:類固醇激素(Steroid)與細胞內受體結合後,hsp90 脫離,受體-配體複合物進入細胞核調控基因轉錄
機制二:配體門控離子通道(Ligand-Gated Ion Channels)#
膜蛋白受體本身就是離子通道,配體結合後通道開啟:
- 效應極快(毫秒),為快速突觸傳遞的基礎
- 例如:乙醯膽鹼(acetylcholine)結合菸鹼型受體(nicotinic receptor, nAChR)→ Na⁺/K⁺ 通道開啟 → 去極化
- 苯二氮平類藥物(benzodiazepines)增強 GABA_A 受體(Cl⁻ 通道)功能
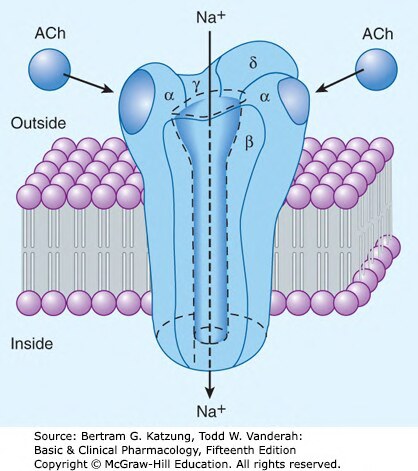
Figure 2-9:菸鹼型乙醯膽鹼受體(nAChR)的五聚體結構,兩個 α 次單元結合 ACh 後開啟中央 Na⁺ 通道
機制三:G 蛋白偶合受體(G Protein-Coupled Receptors, GPCRs)#
GPCRs 是藥物最重要的靶點之一,具有七個跨膜螺旋區域(7TM):
- 激動劑結合 GPCR
- GPCR 構型改變,活化三聚體 G 蛋白(Gα、Gβ、Gγ)
- Gα 與 GTP 結合,與 Gβγ 解離
- 活化的 Gα 或 Gβγ 調控下游效應子(effectors)
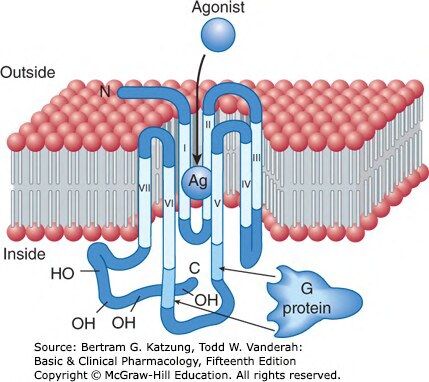
Figure 2-11:GPCR 的七個跨膜螺旋(I–VII)結構,激動劑結合於胞外端,胞內端偶合 G 蛋白
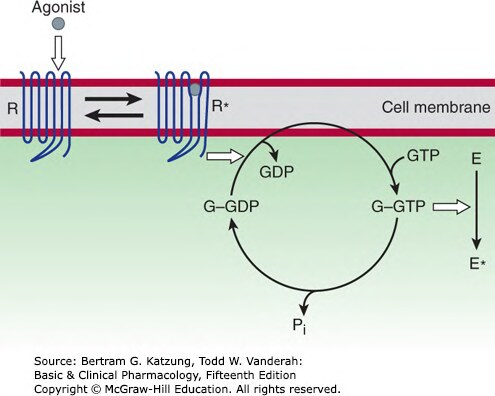
Figure 2-10:G 蛋白活化循環——激動劑結合使受體(R)轉變為 R*,促進 G 蛋白 GDP→GTP 交換,活化下游效應子(E*)
不同 G 蛋白亞型產生不同的細胞內訊號:
| G 蛋白類型 | 效應子 | 第二傳訊者 |
|---|---|---|
| Gs | 活化腺苷環化酶(adenylyl cyclase) | ↑ cAMP |
| Gi | 抑制腺苷環化酶 | ↓ cAMP |
| Gq | 活化磷脂酶 C(PLC) | ↑ IP₃、DAG,→ ↑ Ca²⁺ |
| G₁₂/₁₃ | 活化 Rho GEF | 細胞骨架調控 |
機制四:受體酪氨酸激酶(Receptor Tyrosine Kinases, RTKs)#
- 配體(如生長因子)結合後,受體二聚化(dimerization)
- 胞內激酶域自身磷酸化(autophosphorylation)
- 啟動 Ras-MAPK、PI3K-Akt 等訊號路徑
- 例如:胰島素(insulin)受體、表皮生長因子(EGF)受體
- 靶向 RTK 的藥物:伊馬替尼(imatinib)、曲妥珠單抗(trastuzumab)
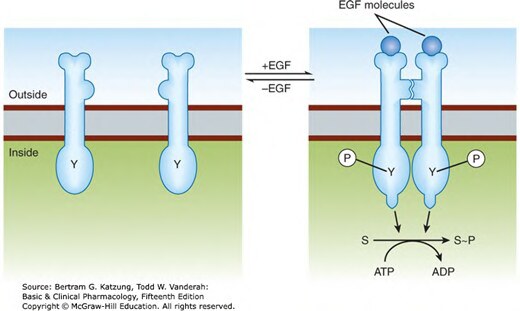
Figure 2-7:EGF 受體酪氨酸激酶——EGF 結合後受體二聚化,胞內酪氨酸域自身磷酸化(Y→Y-P),催化下游受質磷酸化
機制五:細胞激素受體(Cytokine Receptors)/ JAK-STAT 路徑#
- 受體本身無酶活性,但與胞內 JAK(Janus kinase)偶合
- 配體結合 → JAK 活化 → STAT 磷酸化 → 進入細胞核調控基因表現
- JAK 抑制劑(JAK inhibitors):托法替尼(tofacitinib)用於類風濕性關節炎
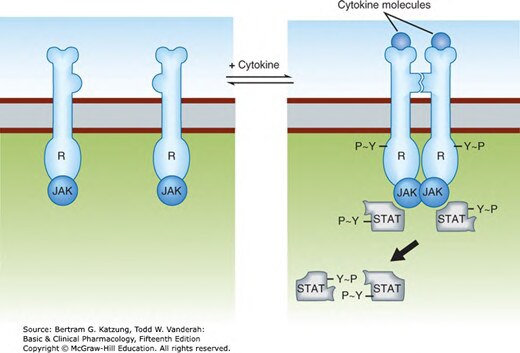
Figure 2-8:細胞激素受體 JAK-STAT 路徑——細胞激素結合後受體二聚化,JAK 相互磷酸化,STAT 磷酸化後形成二聚體入核調控基因
第二傳訊者系統#
cAMP 路徑#
腺苷環化酶被 Gs 活化後,催化 ATP 轉換為環磷酸腺苷(cyclic AMP, cAMP):
- cAMP 活化蛋白激酶 A(PKA)
- PKA 磷酸化多種蛋白質,調控代謝、心肌收縮力、平滑肌舒張等
- 磷酸二酯酶(phosphodiesterase, PDE) 分解 cAMP;PDE 抑制劑(如茶鹼 theophylline、西地那非 sildenafil)可延長 cAMP/cGMP 效應
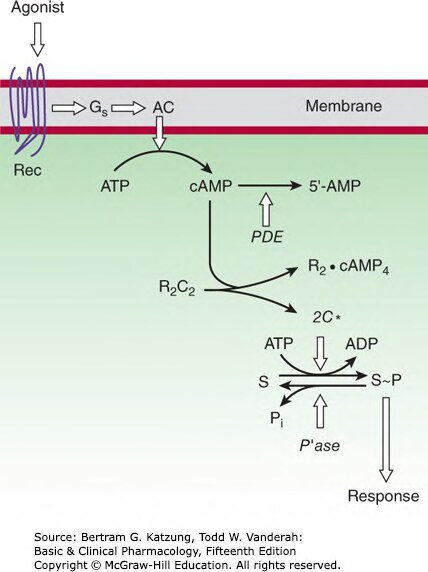
Figure 2-13:Gs-腺苷環化酶-cAMP-PKA 訊號路徑——受體活化 Gs,促進 ATP 轉化為 cAMP,PKA 磷酸化受質蛋白產生細胞反應
磷酸肌醇(Phosphoinositide)與鈣離子路徑#
Gq 活化磷脂酶 C-β(PLCβ),水解 PIP₂ 為:
- IP₃(三磷酸肌醇):作用於內質網 IP₃ 受體 → 釋放 Ca²⁺
- DAG(二酯酰甘油):留在細胞膜,與 Ca²⁺ 共同活化蛋白激酶 C(PKC)
Ca²⁺ 結合鈣調蛋白(calmodulin) 後,調控多種激酶(如 MLCK、CaM kinase)。
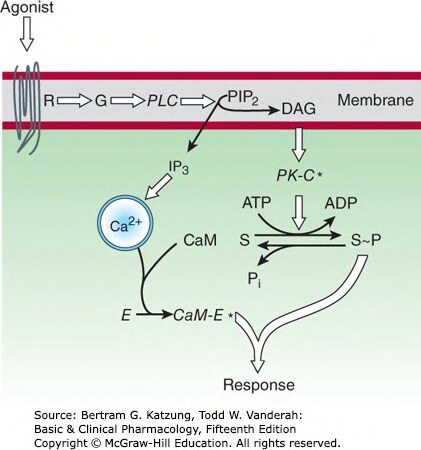
Figure 2-14:Gq-PLC-IP₃/DAG 訊號路徑——PLC 水解 PIP₂ 產生 IP₃(釋放 Ca²⁺)和 DAG(活化 PKC),共同磷酸化受質蛋白
cGMP 路徑#
環磷酸鳥苷(cyclic GMP, cGMP) 由鳥苷環化酶(guanylyl cyclase)合成:
- 一氧化氮(nitric oxide, NO)活化可溶性鳥苷環化酶 → ↑ cGMP → 平滑肌舒張(血管擴張)
- 硝酸甘油(nitroglycerin)在體內釋放 NO 而發揮作用
- 西地那非(sildenafil,威而鋼)抑制 PDE5(分解 cGMP),增強 NO 介導的陰莖海綿體舒張
受體調控:減敏與增敏#
長期暴露於激動劑或拮抗劑會使受體數量或功能改變:
減敏(Desensitization)/ 向下調控(Down-regulation)#
- 短期減敏:GPCR 激酶(G protein-coupled receptor kinases, GRKs)磷酸化活化的受體 → β-arrestin 結合 → 受體與 G 蛋白解偶聯
- 長期減敏:受體向下調控,細胞表面受體數量減少(內化至胞內)
- 臨床意義:β₂ 腎上腺素受體激動劑(如沙丁胺醇 salbutamol)長期使用後療效降低
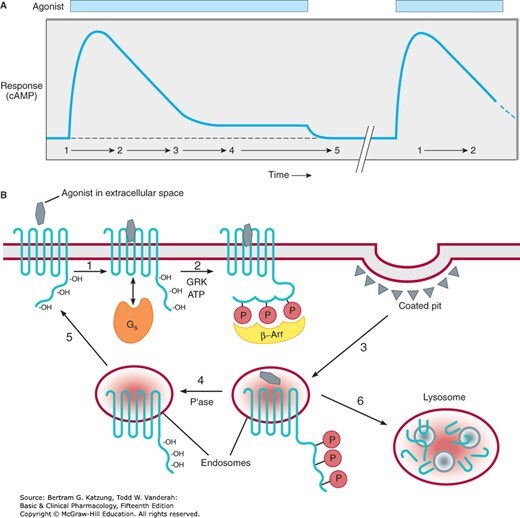
Figure 2-12:GPCR 減敏機制——持續激動劑刺激下,GRK 磷酸化受體(步驟 2)、β-arrestin 結合(3)、clathrin 包裹內化至內體(4–6),或去磷酸化後再循環至細胞表面(5)
增敏(Sensitization)/ 向上調控(Up-regulation)#
- 長期使用拮抗劑 → 受體數量增加、對激動劑敏感性升高
- 突然停藥時出現反彈現象(rebound):如長期使用 β-blocker 後突然停藥,心臟對兒茶酚胺(catecholamines)高度敏感,可能誘發心絞痛甚至心肌梗塞
β 受體阻斷劑不可驟然停藥,應逐漸減量(tapering),避免反彈性高血壓或心絞痛。
藥物劑量-反應關係#
分級反應(Graded Response)#
量效曲線以連續量化指標(如血壓、心率)表示藥物效應隨濃度的變化。
重要參數:
- 效價(potency):以 EC₅₀ 衡量,不決定臨床優劣
- 效能(efficacy):E_max,決定藥物在最嚴重疾病中的有用性
- 治療指數(therapeutic index, TI):LD₅₀(半數致死量)/ ED₅₀(半數有效量),TI 越大越安全
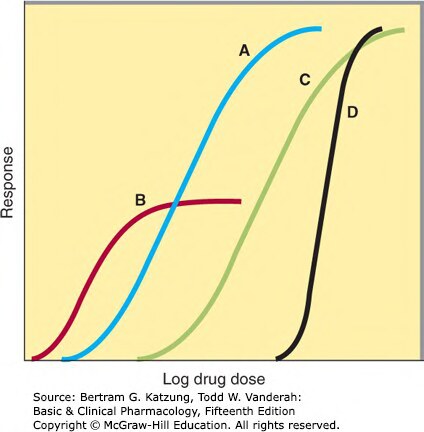
Figure 2-15:多條劑量-反應曲線(A–D)比較不同藥物的效價(曲線左右位置)與效能(曲線最高點),B 曲線代表部分激動劑
最大有效量與毒性#
- 有效劑量範圍(therapeutic window)上限受毒性限制
- 某些藥物(如地高辛 digoxin、鋰鹽 lithium)治療指數窄,需要血中藥物濃度監測(therapeutic drug monitoring, TDM)
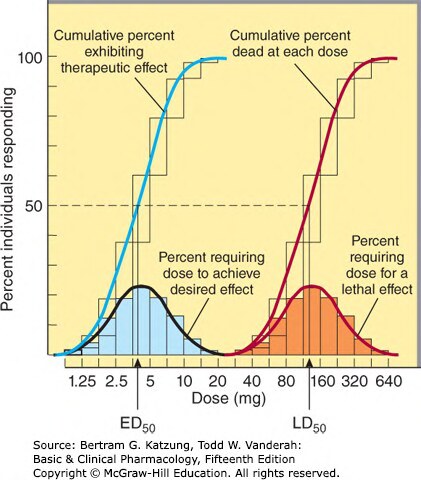
Figure 2-16:群體劑量-反應頻率分布曲線,顯示 ED₅₀(有效劑量)與 LD₅₀(致死劑量)的分布,兩曲線間距反映治療指數大小
藥物反應個體差異#
同樣劑量的藥物,不同個體可能有截然不同的反應,原因包括:
- 藥動學差異:吸收、代謝、排泄速率不同(如 CYP2D6 多型性)
- 藥效學差異:受體數量、敏感性、下游訊號路徑的個體差異
- 疾病狀態:肝腎功能異常改變藥物處置
- 年齡:新生兒(器官未成熟)與老年人(多重用藥、腎功能下降)為高風險族群
- 遺傳藥理學(pharmacogenomics):基因多型性影響藥物代謝酶或受體功能
了解藥物反應個體差異的原因,是實現個人化用藥(personalized medicine)的基礎,也是藥效學與藥動學結合臨床應用的核心。
小結#
藥效學描述藥物與受體結合後如何產生效應,關鍵概念包括:受體的種類與特性、濃度-效應曲線(EC₅₀、E_max)、競爭性與不可逆性拮抗、部分激動劑、五大跨膜訊號傳遞機制(細胞內受體、離子通道、GPCR、RTK、細胞激素受體)、第二傳訊者(cAMP、IP₃/Ca²⁺、cGMP),以及受體調控(減敏/增敏)。理解這些機制有助於預測藥物效應、副作用及個體差異。